

Complete viral RNA genome sequencing of ultra-low copy samples by sequence-independent amplification
- PMID: 22962364
- PMCID: PMC3592391
- DOI: 10.1093/nar/gks794
Complete viral RNA genome sequencing of ultra-low copy samples by sequence-independent amplification
Abstract
RNA viruses are the causative agents for AIDS, influenza, SARS, and other serious health threats. Development of rapid and broadly applicable methods for complete viral genome sequencing is highly desirable to fully understand all aspects of these infectious agents as well as for surveillance of viral pandemic threats and emerging pathogens. However, traditional viral detection methods rely on prior sequence or antigen knowledge. In this study, we describe sequence-independent amplification for samples containing ultra-low amounts of viral RNA coupled with Illumina sequencing and de novo assembly optimized for viral genomes. With 5 million reads, we capture 96 to 100% of the viral protein coding region of HIV, respiratory syncytial and West Nile viral samples from as little as 100 copies of viral RNA. The methods presented here are scalable to large numbers of samples and capable of generating full or near full length viral genomes from clone and clinical samples with low amounts of viral RNA, without prior sequence information and in the presence of substantial host contamination.
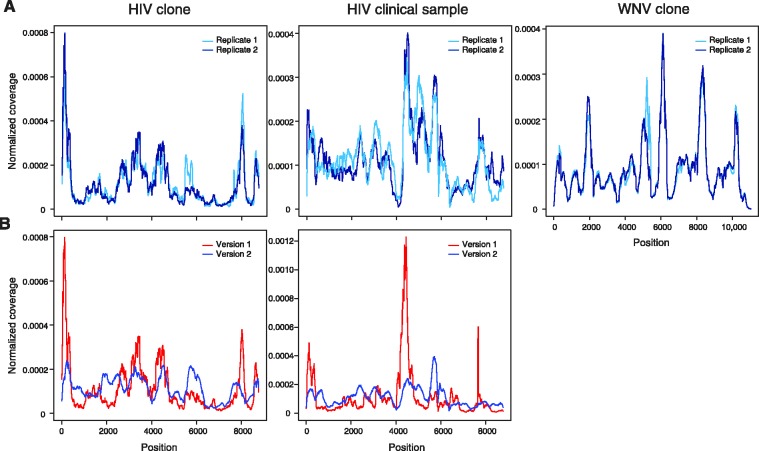
Figures
Similar articles
-
Improved strategies for sequence-independent amplification and sequencing of viral double-stranded RNA genomes.J Gen Virol. 2009 Jun;90(Pt 6):1423-1432. doi: 10.1099/vir.0.009381-0. Epub 2009 Mar 4. J Gen Virol. 2009. PMID: 19264638
-
Viral genome sequencing by random priming methods.BMC Genomics. 2008 Jan 7;9:5. doi: 10.1186/1471-2164-9-5. BMC Genomics. 2008. PMID: 18179705 Free PMC article.
-
Improved assembly procedure of viral RNA genomes amplified with Phi29 polymerase from new generation sequencing data.Biol Res. 2016 Sep 7;49(1):39. doi: 10.1186/s40659-016-0099-y. Biol Res. 2016. PMID: 27605096 Free PMC article.
-
Tracheal aspirate as a substrate for polymerase chain reaction detection of viral genome in childhood pneumonia and myocarditis.Circulation. 1999 Apr 20;99(15):2011-8. doi: 10.1161/01.cir.99.15.2011. Circulation. 1999. PMID: 10209006
-
[Establishment of a system for determination of emerging infectious diseases (RDV method), and its application].Uirusu. 2007 Dec;57(2):217-25. doi: 10.2222/jsv.57.217. Uirusu. 2007. PMID: 18357760 Review. Japanese.
Cited by
-
drVM: a new tool for efficient genome assembly of known eukaryotic viruses from metagenomes.Gigascience. 2017 Feb 1;6(2):1-10. doi: 10.1093/gigascience/gix003. Gigascience. 2017. PMID: 28369462 Free PMC article.
-
Single primer isothermal amplification (SPIA) combined with next generation sequencing provides complete bovine coronavirus genome coverage and higher sequence depth compared to sequence-independent single primer amplification (SISPA).PLoS One. 2017 Nov 7;12(11):e0187780. doi: 10.1371/journal.pone.0187780. eCollection 2017. PLoS One. 2017. PMID: 29112950 Free PMC article.
-
RNA Sequencing Data Sets and Their Whole-Genome Sequence Assembly of Dengue Virus from Three Serial Passages in Vero Cells.Microbiol Resour Announc. 2021 Apr 29;10(17):e00145-21. doi: 10.1128/MRA.00145-21. Microbiol Resour Announc. 2021. PMID: 33927030 Free PMC article.
-
Unbiased Deep Sequencing of RNA Viruses from Clinical Samples.J Vis Exp. 2016 Jul 2;(113):54117. doi: 10.3791/54117. J Vis Exp. 2016. PMID: 27403729 Free PMC article.
-
Deep sequencing of protease inhibitor resistant HIV patient isolates reveals patterns of correlated mutations in Gag and protease.PLoS Comput Biol. 2015 Apr 20;11(4):e1004249. doi: 10.1371/journal.pcbi.1004249. eCollection 2015 Apr. PLoS Comput Biol. 2015. PMID: 25894830 Free PMC article.
References
-
- Simen BB, Simons JF, Hullsiek KH, Novak RM, Macarthur RD, Baxter JD, Huang C, Lubeski C, Turenchalk GS, Braverman MS, et al. Low-abundance drug-resistant viral variants in chronically HIV-infected, antiretroviral treatment-naive patients significantly impact treatment outcomes. J. Infect. Dis. 2009;199:693–701. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
