

Evolution of gene neighborhoods within reconciled phylogenies
- PMID: 22962456
- PMCID: PMC3436801
- DOI: 10.1093/bioinformatics/bts374
Evolution of gene neighborhoods within reconciled phylogenies
Abstract
Motivation: Most models of genome evolution integrating gene duplications, losses and chromosomal rearrangements are computationally intract able, even when comparing only two genomes. This prevents large-scale studies that consider different types of genome structural variations.
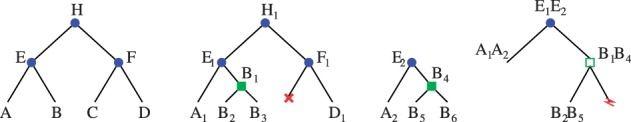
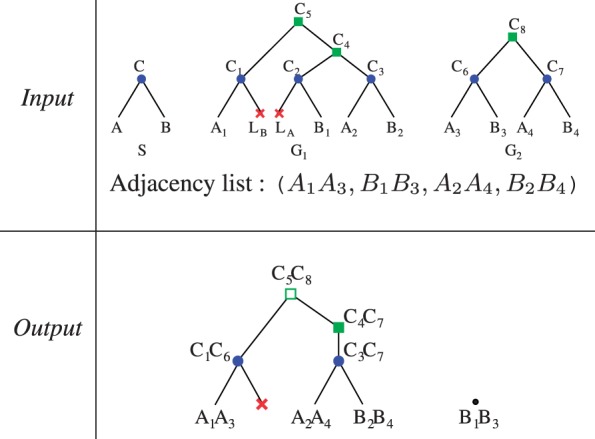
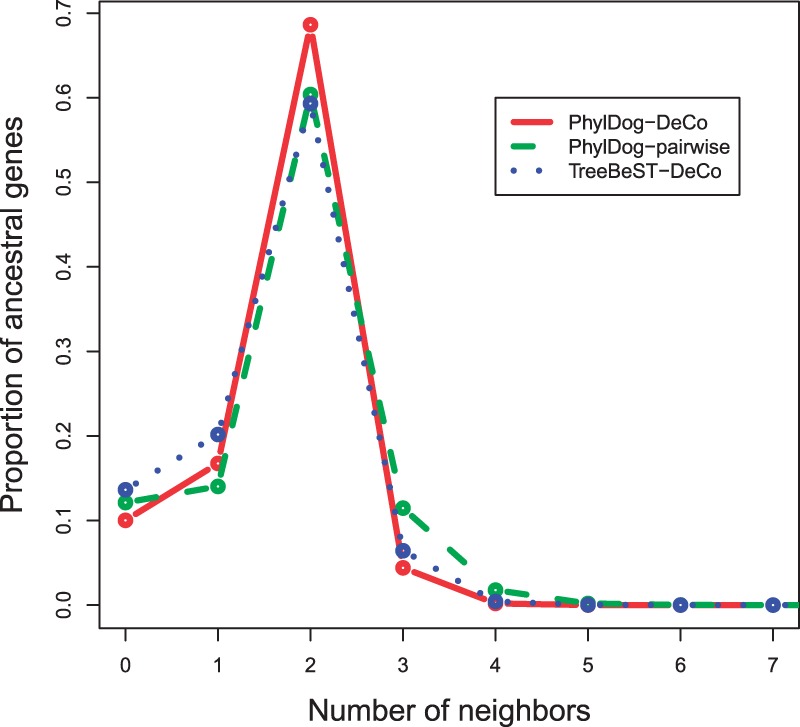
Results: We define an 'adjacency phylogenetic tree' that describes the evolution of an adjacency, a neighborhood relation between two genes, by speciation, duplication or loss of one or both genes, and rearrangement. We describe an algorithm that, given a species tree and a set of gene trees where the leaves are connected by adjacencies, computes an adjacency forest that minimizes the number of gains and breakages of adjacencies (caused by rearrangements) and runs in polynomial time. We use this algorithm to reconstruct contiguous regions of mammalian and plant ancestral genomes in a few minutes for a dozen species and several thousand genes. We show that this method yields reduced conflict between ancestral adjacencies. We detect duplications involving several genes and compare the different modes of evolution between phyla and among lineages.
Availability: C++ implementation using BIO++ package, available upon request to Sèverine Bérard.
Contact: Severine.Berard@cirad.fr or Eric.Tannier@inria.fr
Supplementary information: Supplementary material is available at Bioinformatics online.
Figures

References
-
- Bertrand D., et al. Proceedings of WABI‘10, Algorithms in Bioinformatics. Berlin Heidelberg: Lecture Notes in Bioinformatics, Springer; 2010. Reconstruction of ancestral genome subject to whole genome duplication, speciation, rearrangement and loss; pp. 78–89.
-
- Chauve C., et al. Yeast ancestral genome reconstructions: the possibilities of computational methods II. J. Comput. Biol. 2010;17:1097–1112. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
