

Weak selection and protein evolution
- PMID: 22964835
- PMCID: PMC3430532
- DOI: 10.1534/genetics.112.140178
Weak selection and protein evolution
Abstract
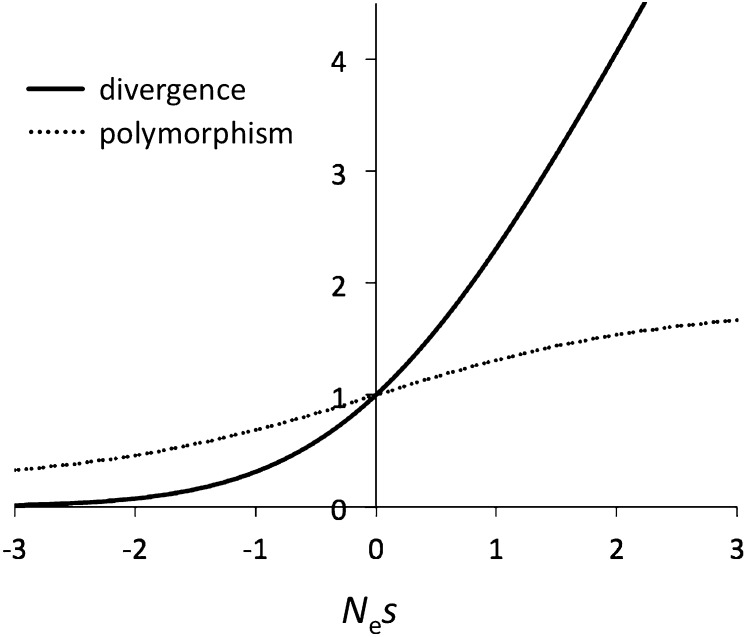
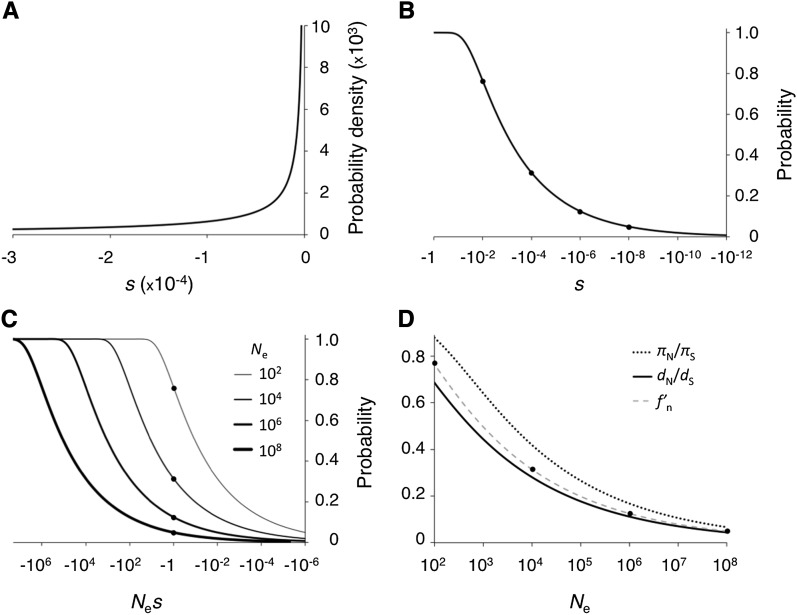
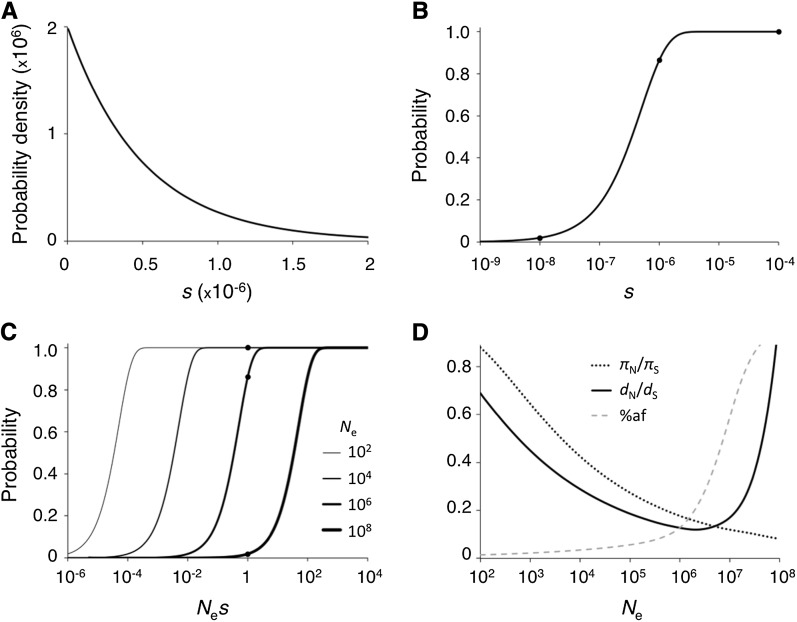
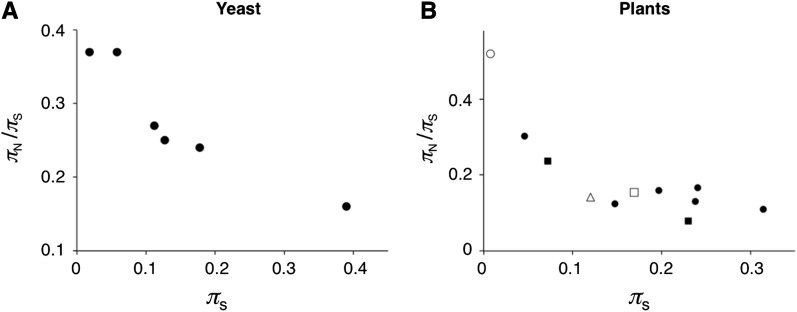
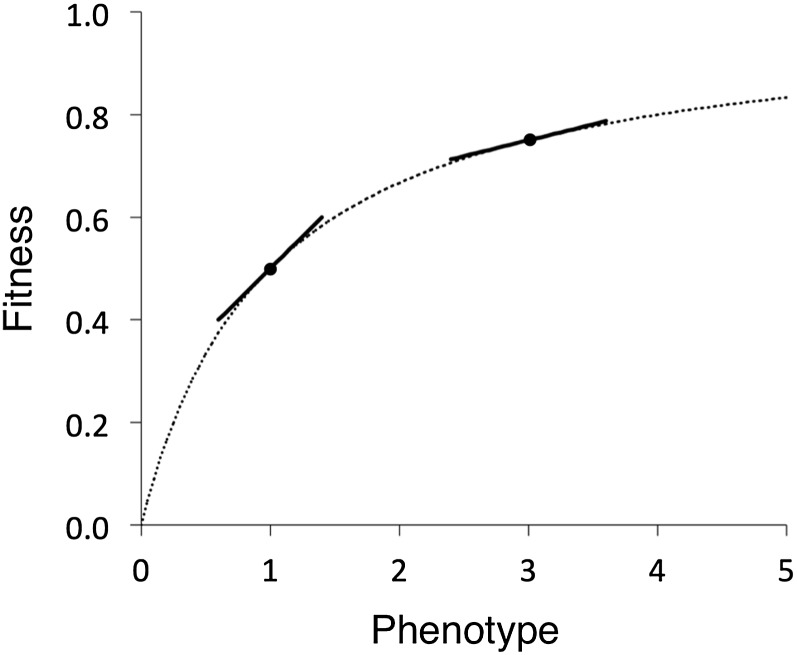
The "nearly neutral" theory of molecular evolution proposes that many features of genomes arise from the interaction of three weak evolutionary forces: mutation, genetic drift, and natural selection acting at its limit of efficacy. Such forces generally have little impact on allele frequencies within populations from generation to generation but can have substantial effects on long-term evolution. The evolutionary dynamics of weakly selected mutations are highly sensitive to population size, and near neutrality was initially proposed as an adjustment to the neutral theory to account for general patterns in available protein and DNA variation data. Here, we review the motivation for the nearly neutral theory, discuss the structure of the model and its predictions, and evaluate current empirical support for interactions among weak evolutionary forces in protein evolution. Near neutrality may be a prevalent mode of evolution across a range of functional categories of mutations and taxa. However, multiple evolutionary mechanisms (including adaptive evolution, linked selection, changes in fitness-effect distributions, and weak selection) can often explain the same patterns of genome variation. Strong parameter sensitivity remains a limitation of the nearly neutral model, and we discuss concave fitness functions as a plausible underlying basis for weak selection.
Figures
References
-
- Abbot P., Moran N. A., 2002. Extremely low levels of genetic polymorphism in endosymbionts (Buchnera) of aphids (Pemphigus). Mol. Ecol. 11: 2649–2660 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
