

Modeling of epigenome dynamics identifies transcription factors that mediate Polycomb targeting
- PMID: 22964890
- PMCID: PMC3530684
- DOI: 10.1101/gr.142661.112
Modeling of epigenome dynamics identifies transcription factors that mediate Polycomb targeting
Abstract
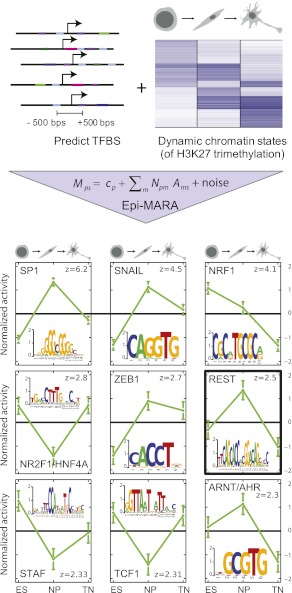
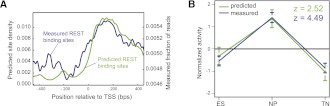
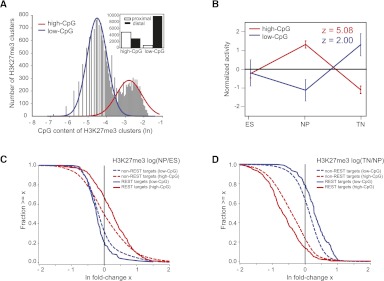
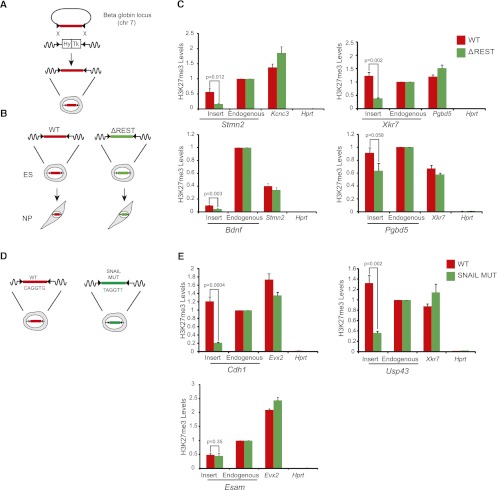
Although changes in chromatin are integral to transcriptional reprogramming during cellular differentiation, it is currently unclear how chromatin modifications are targeted to specific loci. To systematically identify transcription factors (TFs) that can direct chromatin changes during cell fate decisions, we model the relationship between genome-wide dynamics of chromatin marks and the local occurrence of computationally predicted TF binding sites. By applying this computational approach to a time course of Polycomb-mediated H3K27me3 marks during neuronal differentiation of murine stem cells, we identify several motifs that likely regulate the dynamics of this chromatin mark. Among these, the sites bound by REST and by the SNAIL family of TFs are predicted to transiently recruit H3K27me3 in neuronal progenitors. We validate these predictions experimentally and show that absence of REST indeed causes loss of H3K27me3 at target promoters in trans, specifically at the neuronal progenitor state. Moreover, using targeted transgenic insertion, we show that promoter fragments containing REST or SNAIL binding sites are sufficient to recruit H3K27me3 in cis, while deletion of these sites results in loss of H3K27me3. These findings illustrate that the occurrence of TF binding sites can determine chromatin dynamics. Local determination of Polycomb activity by REST and SNAIL motifs exemplifies such TF based regulation of chromatin. Furthermore, our results show that key TFs can be identified ab initio through computational modeling of epigenome data sets using a modeling approach that we make readily accessible.
Figures

References
-
- Abbott A 2011. Europe to map the human epigenome. Nature 477: 518 doi: 10.1038/477518a - PubMed
-
- Arnold P, Erb I, Pachkov M, Molina N, van Nimwegen E 2012. MotEvo: Integrated Bayesian probabilistic methods for inferring regulatory sites and motifs on multiple alignments of DNA sequences. Bioinformatics 28: 487–494 - PubMed
-
- Barrera LO, Ren B 2006. The transcriptional regulatory code of eukaryotic cells—insights from genome-wide analysis of chromatin organization and transcription factor binding. Curr Opin Cell Biol 18: 291–298 - PubMed
-
- Beer MA, Tavazoie S 2004. Predicting gene expression from sequence. Cell 117: 185–198 - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous