

Inverted low-copy repeats and genome instability--a genome-wide analysis
- PMID: 22965494
- PMCID: PMC3738003
- DOI: 10.1002/humu.22217
Inverted low-copy repeats and genome instability--a genome-wide analysis
Abstract
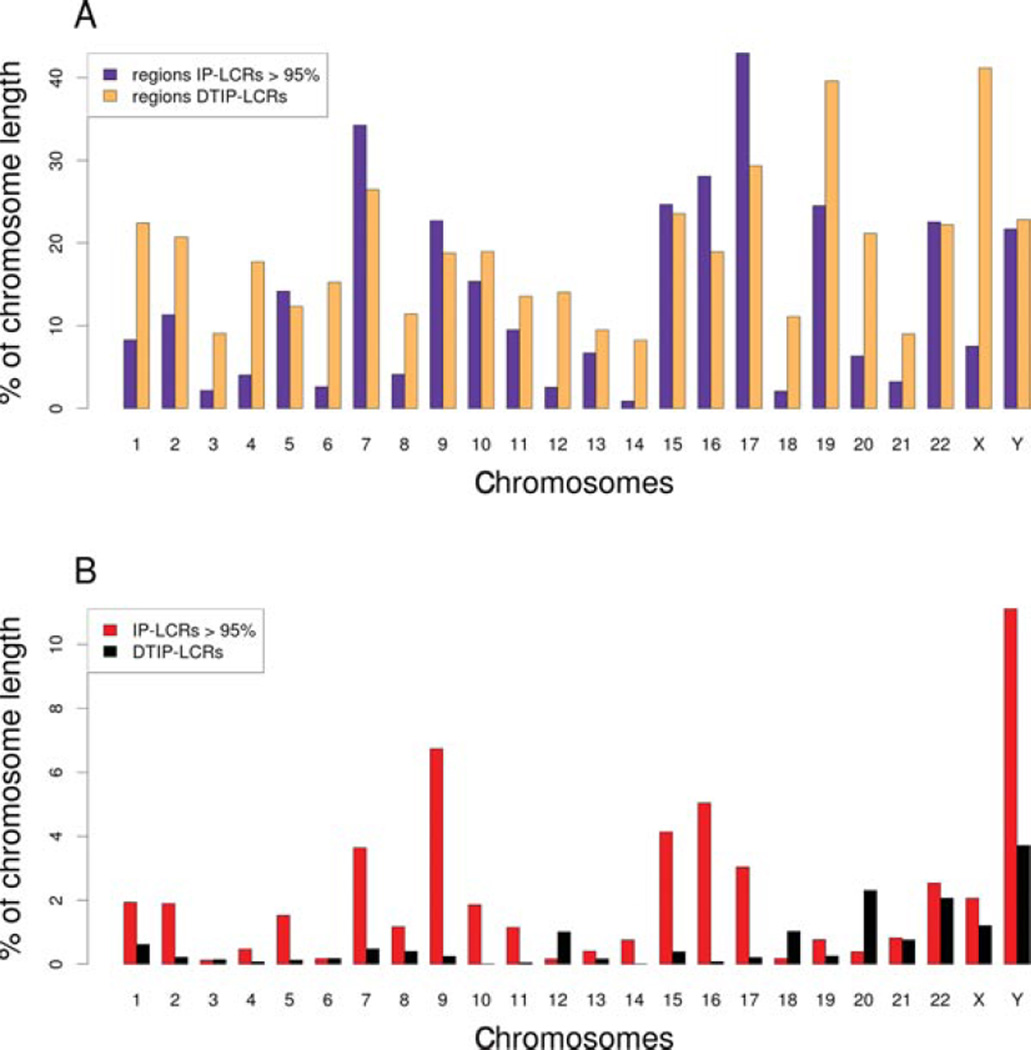
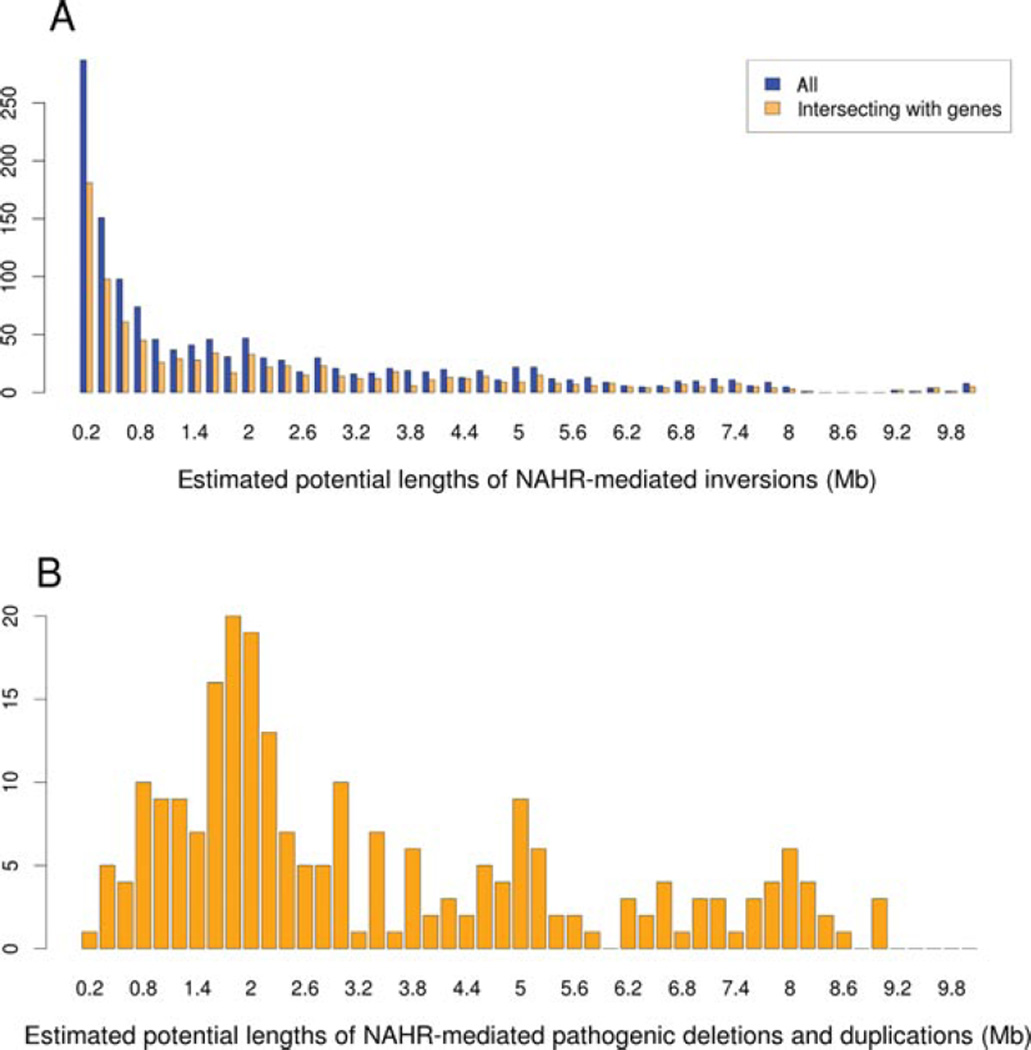
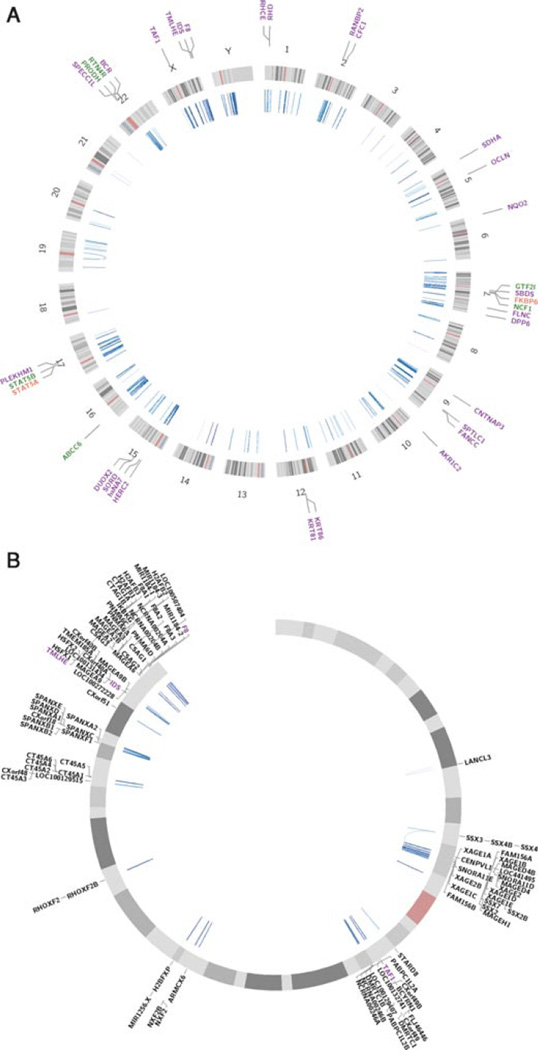
Inverse paralogous low-copy repeats (IP-LCRs) can cause genome instability by nonallelic homologous recombination (NAHR)-mediated balanced inversions. When disrupting a dosage-sensitive gene(s), balanced inversions can lead to abnormal phenotypes. We delineated the genome-wide distribution of IP-LCRs >1 kB in size with >95% sequence identity and mapped the genes, potentially intersected by an inversion, that overlap at least one of the IP-LCRs. Remarkably, our results show that 12.0% of the human genome is potentially susceptible to such inversions and 942 genes, 99 of which are on the X chromosome, are predicted to be disrupted secondary to such an inversion! In addition, IP-LCRs larger than 800 bp with at least 98% sequence identity (duplication/triplication facilitating IP-LCRs, DTIP-LCRs) were recently implicated in the formation of complex genomic rearrangements with a duplication-inverted triplication-duplication (DUP-TRP/INV-DUP) structure by a replication-based mechanism involving a template switch between such inverted repeats. We identified 1,551 DTIP-LCRs that could facilitate DUP-TRP/INV-DUP formation. Remarkably, 1,445 disease-associated genes are at risk of undergoing copy-number gain as they map to genomic intervals susceptible to the formation of DUP-TRP/INV-DUP complex rearrangements. We implicate inverted LCRs as a human genome architectural feature that could potentially be responsible for genomic instability associated with many human disease traits.
© 2012 Wiley Periodicals, Inc.
Figures

References
-
- Alders M, Koopmann TT, Christiaans I, Postema PG, Beekman L, Tanck MWT, Zeppenfeld K, Loh P, Koch KT, Demolombe S, Mannens MM, Bezzina CR, et al. Haplotype-sharing analysis implicates chromosome 7q36 harboring DPP6 in familial idiopathic ventricular fibrillation. Am J Hum Genet. 2009;84:468–476. - PMC - PubMed
-
- Aradhya S, Bardaro T, Galgóczy P, Yamagata T, Esposito T, Patlan H, Ciccodicola A, Munnich A, Kenwrick S, Platzer M, D’Urso M, Nelson DL. Multiple pathogenic and benign genomic rearrangements occur at a 35 kB duplication involving the NEMO and LAGE2 genes. Hum Mol Genet. 2001;10:2557–2567. - PubMed
-
- Auclair J, Leroux D, Desseigne F, Lasset C, Saurin JC, Joly MO, Pinson S, Xu XL, Montmain G, Ruano E, Navarro C, Puisieux A, et al. Novel biallelic mutations in MSH6 and PMS2 genes: gene conversion as a likely cause of PMS2 gene inactivation. Hum Mutat. 2007;28:1084–1090. - PubMed
-
- Bagnall RD, Waseem N, Green PM, Giannelli F. Recurrent inversion breaking intron 1 of the factor VIII gene is a frequent cause of severe hemophilia A. Blood. 2002;99:168–174. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
