

Antigen and substrate withdrawal in the management of autoimmune thrombotic disorders
- PMID: 22966172
- PMCID: PMC3501713
- DOI: 10.1182/blood-2012-06-389445
Antigen and substrate withdrawal in the management of autoimmune thrombotic disorders
Abstract
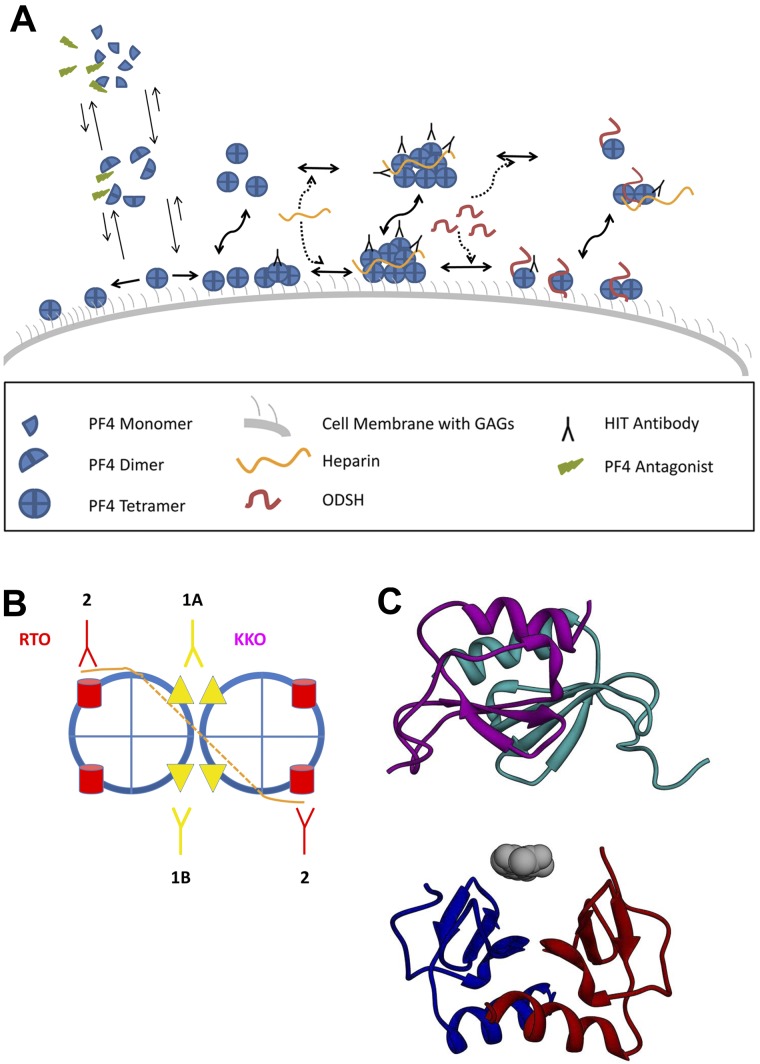
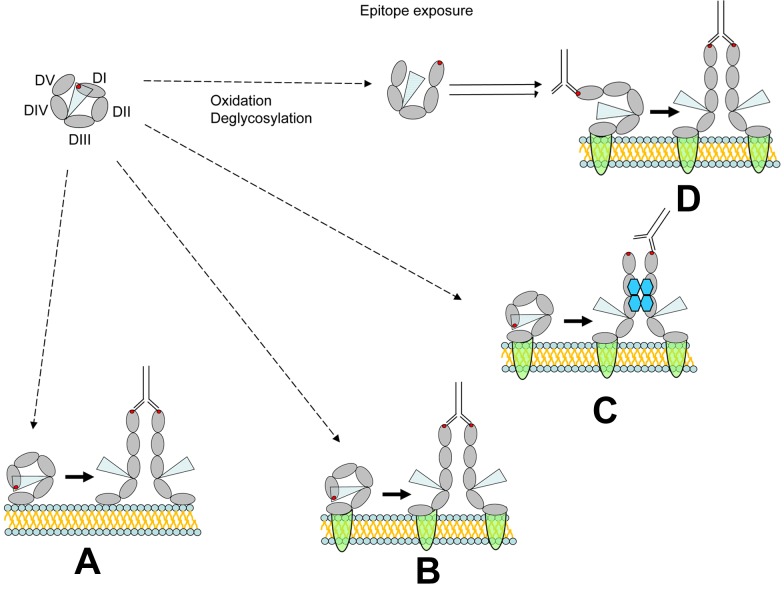
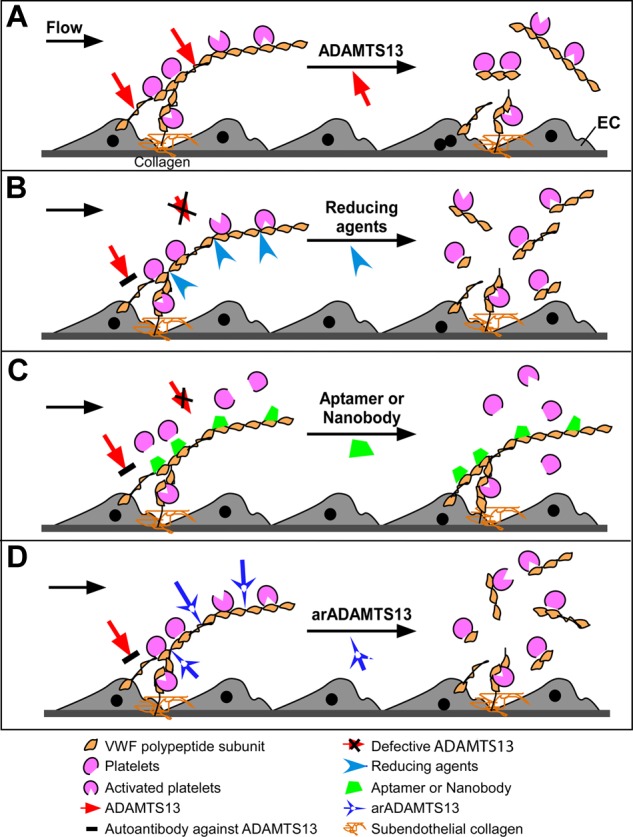
Prevailing approaches to manage autoimmune thrombotic disorders, such as heparin-induced thrombocytopenia, antiphospholipid syndrome and thrombotic thrombocytopenic purpura, include immunosuppression and systemic anticoagulation, though neither provides optimal outcome for many patients. A different approach is suggested by the concurrence of autoantibodies and their antigenic targets in the absence of clinical disease, such as platelet factor 4 in heparin-induced thrombocytopenia and β(2)-glycoprotein-I (β(2)GPI) in antiphospholipid syndrome. The presence of autoantibodies in the absence of disease suggests that conformational changes or other alterations in endogenous protein autoantigens are required for recognition by pathogenic autoantibodies. In thrombotic thrombocytopenic purpura, the clinical impact of ADAMTS13 deficiency caused by autoantibodies likely depends on the balance between residual antigen, that is, enzyme activity, and demand imposed by local genesis of ultralarge multimers of von Willebrand factor. A corollary of these concepts is that disrupting platelet factor 4 and β(2)GPI conformation (or ultralarge multimer of von Willebrand factor oligomerization or function) might provide a disease-targeted approach to prevent thrombosis without systemic anticoagulation or immunosuppression. Validation of this approach requires a deeper understanding of how seemingly normal host proteins become antigenic or undergo changes that increase antibody avidity, and how they can be altered to retain adaptive functions while shedding epitopes prone to elicit harmful autoimmunity.
Figures
Similar articles
-
Thrombotic thrombocytopenic purpura and autoimmunity: a tale of shadows and suspects.Haematologica. 1999 Mar;84(3):260-9. Haematologica. 1999. PMID: 10189393 Review.
-
beta2-Glycoprotein I inhibits von Willebrand factor dependent platelet adhesion and aggregation.Blood. 2007 Sep 1;110(5):1483-91. doi: 10.1182/blood-2006-10-053199. Epub 2007 May 8. Blood. 2007. PMID: 17488878 Clinical Trial.
-
Plasmin cleavage of von Willebrand factor as an emergency bypass for ADAMTS13 deficiency in thrombotic microangiopathy.Circulation. 2014 Mar 25;129(12):1320-31. doi: 10.1161/CIRCULATIONAHA.113.006727. Epub 2014 Jan 21. Circulation. 2014. PMID: 24449821
-
ADAMTS13 and anti-ADAMTS13 autoantibodies in thrombotic thrombocytopenic purpura - current perspectives and new treatment strategies.Expert Rev Hematol. 2016;9(2):209-21. doi: 10.1586/17474086.2016.1122515. Epub 2015 Dec 8. Expert Rev Hematol. 2016. PMID: 26581428 Review.
-
ADAMTS13 and anti-ADAMTS13 antibodies as markers for recurrence of acquired thrombotic thrombocytopenic purpura during remission.Haematologica. 2008 Feb;93(2):232-9. doi: 10.3324/haematol.11739. Epub 2008 Jan 26. Haematologica. 2008. PMID: 18223285
Cited by
-
ADAMTS13, TTP and Beyond.Hereditary Genet. 2013;2(1):e104. doi: 10.4172/2161-1041.1000e104. Hereditary Genet. 2013. PMID: 24790789 Free PMC article. No abstract available.
-
Platelet immunology of patients with hemophilia.Am J Transl Res. 2021 Aug 15;13(8):9831-9838. eCollection 2021. Am J Transl Res. 2021. PMID: 34540118 Free PMC article.
-
Emphasis on the Role of PF4 in the Incidence, Pathophysiology and Treatment of Heparin Induced Thrombocytopenia.Thromb J. 2013 Apr 5;11(1):7. doi: 10.1186/1477-9560-11-7. Thromb J. 2013. PMID: 23561460 Free PMC article.
-
Mechanisms of Immunothrombosis in Vaccine-Induced Thrombotic Thrombocytopenia (VITT) Compared to Natural SARS-CoV-2 Infection.J Autoimmun. 2021 Jul;121:102662. doi: 10.1016/j.jaut.2021.102662. Epub 2021 May 19. J Autoimmun. 2021. PMID: 34051613 Free PMC article. Review.
References
-
- Aster RH, Bougie DW. Drug-induced immune thrombocytopenia. N Engl J Med. 2007;357(6):580–587. - PubMed
-
- Cuker A, Coles AJ, Sullivan H, et al. A distinctive form of immune thrombocytopenia in a phase 2 study of alemtuzumab for the treatment of relapsing-remitting multiple sclerosis. Blood. 2011;118(24):6299–6305. - PubMed
-
- Mantadakis E, Farmaki E, Buchanan GR. Thrombocytopenic purpura after measles-mumps-rubella vaccination: a systematic review of the literature and guidance for management. J Pediatr. 2010;156(4):623–628. - PubMed
-
- Cines DB. ITP: time to bug off? Blood. 2007;110:3818–3819.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
