

Protein instability and functional defects caused by mutations of dihydro-orotate dehydrogenase in Miller syndrome patients
- PMID: 22967083
- PMCID: PMC3497730
- DOI: 10.1042/BSR20120046
Protein instability and functional defects caused by mutations of dihydro-orotate dehydrogenase in Miller syndrome patients
Abstract
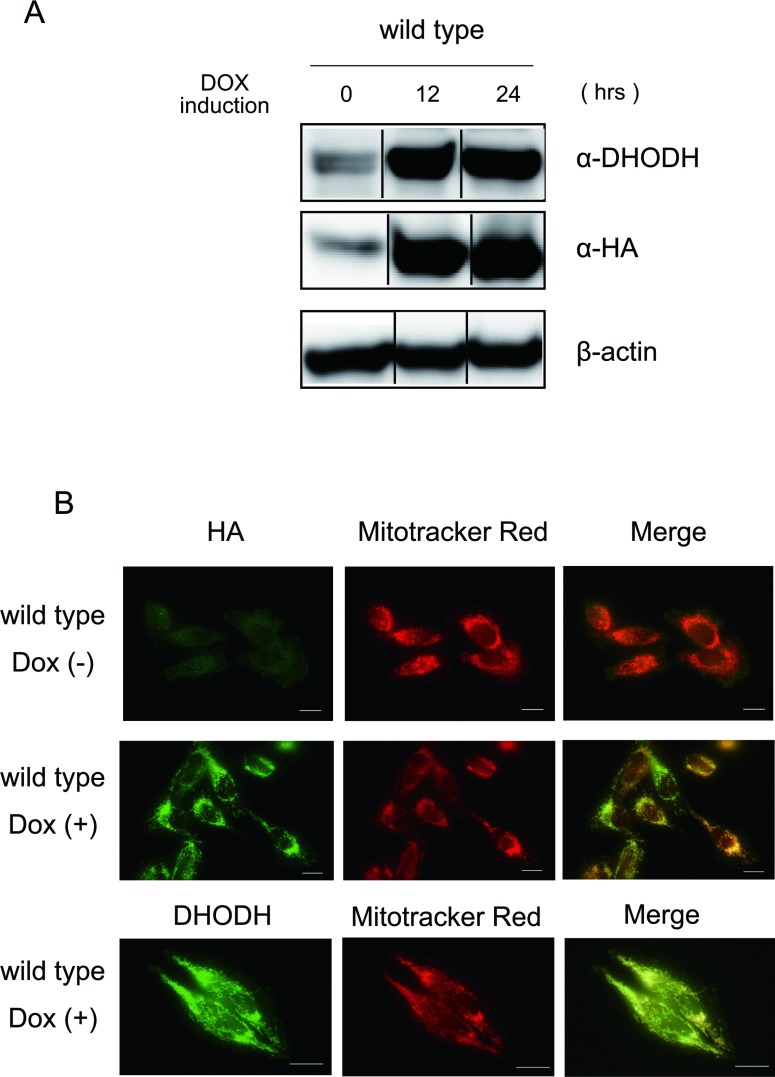
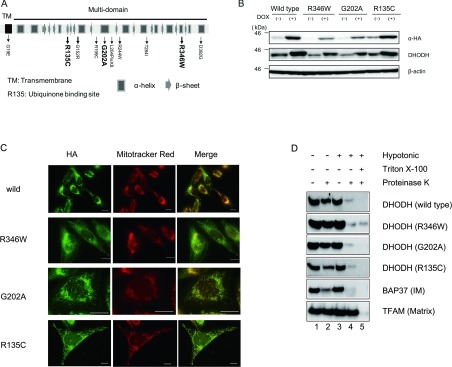
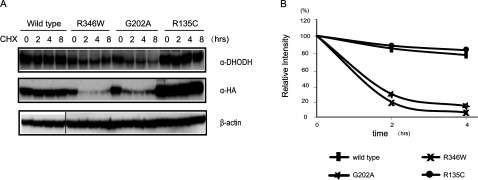
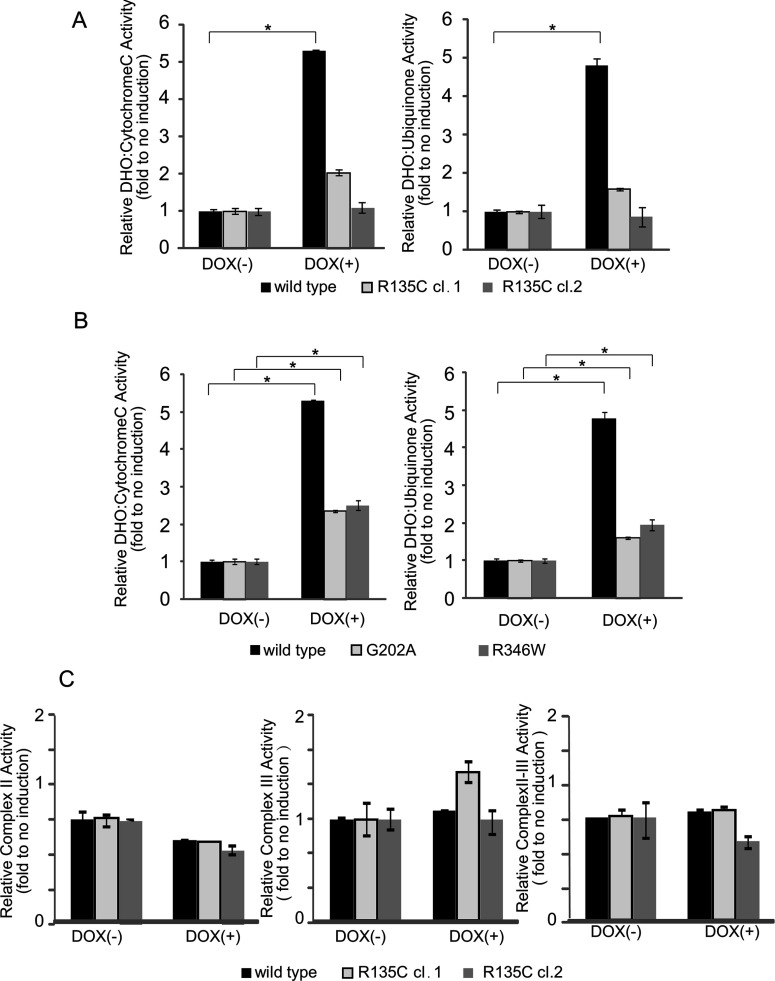
Miller syndrome is a recessive inherited disorder characterized by postaxial acrofacial dysostosis. It is caused by dysfunction of the DHODH (dihydroorotate dehydrogenase) gene, which encodes a key enzyme in the pyrimidine de novo biosynthesis pathway and is localized at mitochondria intermembrane space. We investigated the consequence of three missense mutations, G202A, R346W and R135C of DHODH, which were previously identified in patients with Miller syndrome. First, we established HeLa cell lines stably expressing DHODH with Miller syndrome-causative mutations: G202A, R346W and R135C. These three mutant proteins retained the proper mitochondrial localization based on immunohistochemistry and mitochondrial subfractionation studies. The G202A, R346W DHODH proteins showed reduced protein stability. On the other hand, the third one R135C, in which the mutation lies at the ubiquinone-binding site, was stable but possessed no enzymatic activity. In conclusion, the G202A and R346W mutation causes deficient protein stability, and the R135C mutation does not affect stability but impairs the substrate-induced enzymatic activity, suggesting that impairment of DHODH activity is linked to the Miller syndrome phenotype.
Figures
References
-
- McBride H. M., Neuspiel M., Wasiak S. Mitochondria: more than just a powerhouse. Curr. Biol. 2006;16:R551–R560. - PubMed
-
- DiMauro S., Schon E. A. Mitochondrial disorders in the nervous system. Annu. Rev. Neurosci. 2008;31:91–123. - PubMed
-
- Frenzel M., Rommelspacher H., Sugawa M. D., Dencher N. A. Ageing alters the supramolecular architecture of OxPhos complexes in rat brain cortex. Exp. Gerontol. 2010;45:563–572. - PubMed
-
- Wallace D. C. Mitochondrial DNA mutations in disease and aging. Environ. Mol. Mutagen. 2010;51:440–450. - PubMed
-
- Tuppen H. A., Blakely E. L., Turnbull D. M., Taylor R. W. Mitochondrial DNA mutations and human disease. Biochim. Biophys. Acta. 2010;1797:113–128. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources