

Arginine methylation next to the PY-NLS modulates Transportin binding and nuclear import of FUS
- PMID: 22968170
- PMCID: PMC3501225
- DOI: 10.1038/emboj.2012.261
Arginine methylation next to the PY-NLS modulates Transportin binding and nuclear import of FUS
Abstract
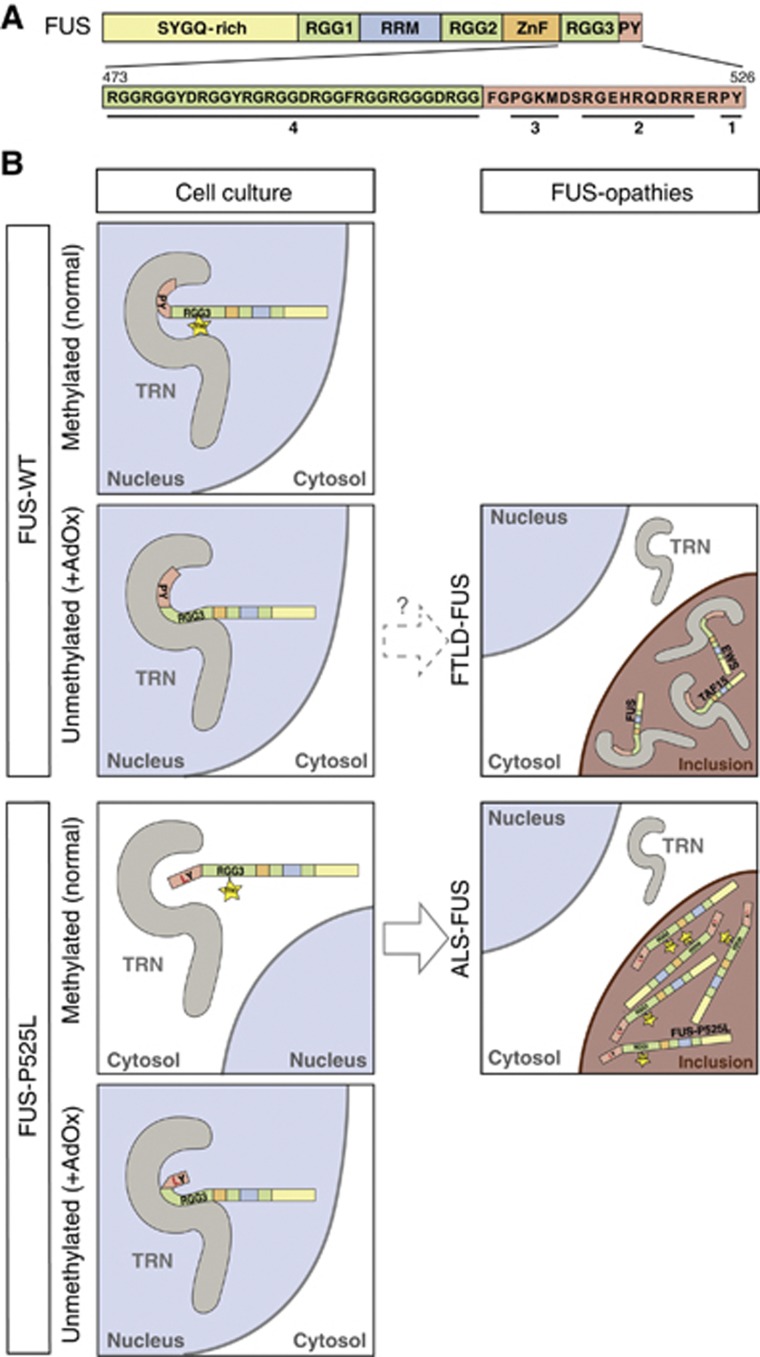
Fused in sarcoma (FUS) is a nuclear protein that carries a proline-tyrosine nuclear localization signal (PY-NLS) and is imported into the nucleus via Transportin (TRN). Defects in nuclear import of FUS have been implicated in neurodegeneration, since mutations in the PY-NLS of FUS cause amyotrophic lateral sclerosis (ALS). Moreover, FUS is deposited in the cytosol in a subset of frontotemporal lobar degeneration (FTLD) patients. Here, we show that arginine methylation modulates nuclear import of FUS via a novel TRN-binding epitope. Chemical or genetic inhibition of arginine methylation restores TRN-mediated nuclear import of ALS-associated FUS mutants. The unmethylated arginine-glycine-glycine domain preceding the PY-NLS interacts with TRN and arginine methylation in this domain reduces TRN binding. Inclusions in ALS-FUS patients contain methylated FUS, while inclusions in FTLD-FUS patients are not methylated. Together with recent findings that FUS co-aggregates with two related proteins of the FET family and TRN in FTLD-FUS but not in ALS-FUS, our study provides evidence that these two diseases may be initiated by distinct pathomechanisms and implicates alterations in arginine methylation in pathogenesis.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures









Comment in
-
The FUS about arginine methylation in ALS and FTLD.EMBO J. 2012 Nov 14;31(22):4249-51. doi: 10.1038/emboj.2012.291. Epub 2012 Oct 19. EMBO J. 2012. PMID: 23085990 Free PMC article.
References
-
- Aoki N, Higashi S, Kawakami I, Kobayashi Z, Hosokawa M, Katsuse O, Togo T, Hirayasu Y, Akiyama H (2012) Localization of fused in sarcoma (FUS) protein to the post-synaptic density in the brain. Acta Neuropathol 124: 383–394 - PubMed
-
- Araya N, Hiraga H, Kako K, Arao Y, Kato S, Fukamizu A (2005) Transcriptional down-regulation through nuclear exclusion of EWS methylated by PRMT1. Biochem Biophys Res Commun 329: 653–660 - PubMed
-
- Bartel RL, Borchardt RT (1984) Effects of adenosine dialdehyde on S-adenosylhomocysteine hydrolase and S-adenosylmethionine-dependent transmethylations in mouse L929 cells. Mol Pharmacol 25: 418–424 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
