

Correlated electrostatic mutations provide a reservoir of stability in HIV protease
- PMID: 22969420
- PMCID: PMC3435258
- DOI: 10.1371/journal.pcbi.1002675
Correlated electrostatic mutations provide a reservoir of stability in HIV protease
Abstract
HIV protease, an aspartyl protease crucial to the life cycle of HIV, is the target of many drug development programs. Though many protease inhibitors are on the market, protease eventually evades these drugs by mutating at a rapid pace and building drug resistance. The drug resistance mutations, called primary mutations, are often destabilizing to the enzyme and this loss of stability has to be compensated for. Using a coarse-grained biophysical energy model together with statistical inference methods, we observe that accessory mutations of charged residues increase protein stability, playing a key role in compensating for destabilizing primary drug resistance mutations. Increased stability is intimately related to correlations between electrostatic mutations - uncorrelated mutations would strongly destabilize the enzyme. Additionally, statistical modeling indicates that the network of correlated electrostatic mutations has a simple topology and has evolved to minimize frustrated interactions. The model's statistical coupling parameters reflect this lack of frustration and strongly distinguish like-charge electrostatic interactions from unlike-charge interactions for [Formula: see text] of the most significantly correlated double mutants. Finally, we demonstrate that our model has considerable predictive power and can be used to predict complex mutation patterns, that have not yet been observed due to finite sample size effects, and which are likely to exist within the larger patient population whose virus has not yet been sequenced.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
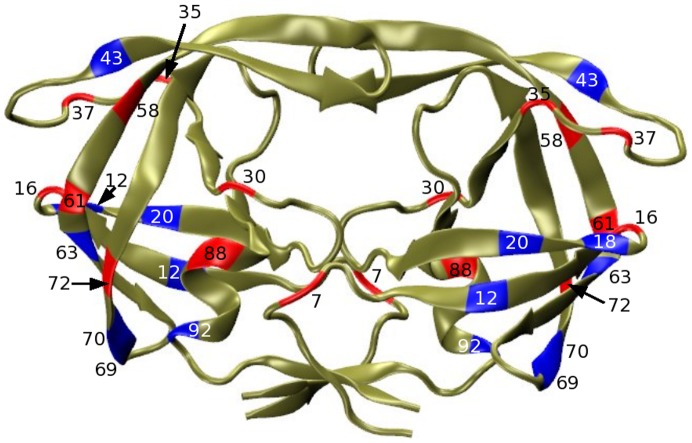
 , where
, where  is the number of sequences with
is the number of sequences with  electrostatic mutations,
electrostatic mutations,  is the probability of the
is the probability of the  th sequence under a given model conditional upon the number of mutations, and
th sequence under a given model conditional upon the number of mutations, and  is its electrostatic folding energy (Equation (1); see Methods). All points are plotted relative to
is its electrostatic folding energy (Equation (1); see Methods). All points are plotted relative to  , the average
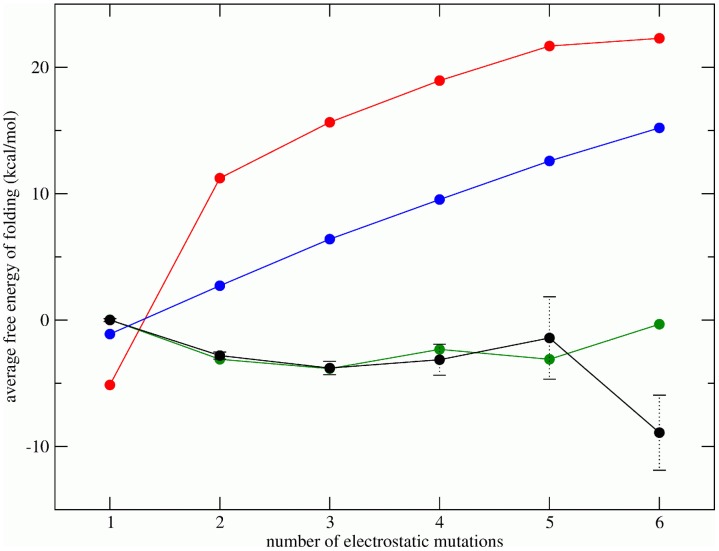
, the average  of observed sequences with one electrostatic mutation. The black curve shows the average energies of observed sequences
of observed sequences with one electrostatic mutation. The black curve shows the average energies of observed sequences  , the red curve represents the average energies of sequences under a model in which each charge state occurs with equal frequency, the blue curve shows the average energies of sequences under a model in which each charge state occurs with frequencies observed in the data, and the green curve represents the average energy of sequences under a pair correlation model which preserves observed pair frequencies. The error bars on the black curve are the standard errors of the mean of observed sequences. Note that
, the red curve represents the average energies of sequences under a model in which each charge state occurs with equal frequency, the blue curve shows the average energies of sequences under a model in which each charge state occurs with frequencies observed in the data, and the green curve represents the average energy of sequences under a pair correlation model which preserves observed pair frequencies. The error bars on the black curve are the standard errors of the mean of observed sequences. Note that  for
for  .
.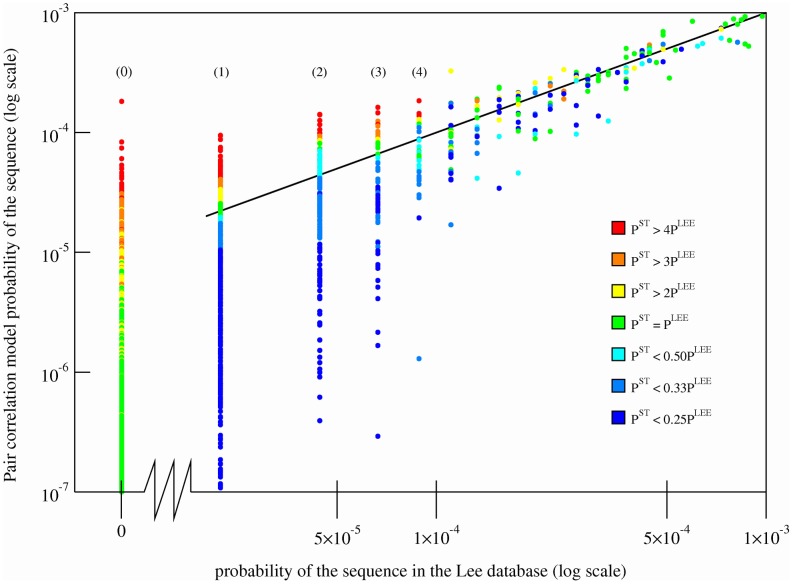
 , predicted using the Bethe approximation, are plotted as a function of the sequence probabilities from the Lee database,
, predicted using the Bethe approximation, are plotted as a function of the sequence probabilities from the Lee database,  . Sequences with a probability of 0 in the Lee database, i.e. unobserved sequences, are plotted to the left of the abscissa break. Every sequence is shaded using a color gradient corresponding to
. Sequences with a probability of 0 in the Lee database, i.e. unobserved sequences, are plotted to the left of the abscissa break. Every sequence is shaded using a color gradient corresponding to  , which represents the number of times the sequence occurs in the Stanford database, relative to its probability in the Lee database. Sequences that occur frequently in the Stanford database as compared to the Lee database have a higher ratio and are shaded red, while the sequences that do not occur as frequently in the Stanford database as compared to the Lee database have a lower ratio and are shaded blue. Sequences that are shaded green have equal probabilities in both databases. Sequences unobserved in the Lee database (leftmost row in the graph), but observed in the Stanford database have a ratio that is artificially set to equal 4, which corresponds to the color red. Unobserved Lee sequences that are also unobserved in the Stanford database are shaded green because
, which represents the number of times the sequence occurs in the Stanford database, relative to its probability in the Lee database. Sequences that occur frequently in the Stanford database as compared to the Lee database have a higher ratio and are shaded red, while the sequences that do not occur as frequently in the Stanford database as compared to the Lee database have a lower ratio and are shaded blue. Sequences that are shaded green have equal probabilities in both databases. Sequences unobserved in the Lee database (leftmost row in the graph), but observed in the Stanford database have a ratio that is artificially set to equal 4, which corresponds to the color red. Unobserved Lee sequences that are also unobserved in the Stanford database are shaded green because  . Sequences with probabilities
. Sequences with probabilities  are shaded according to the average value of
are shaded according to the average value of  for a window of 10 sequences around the sequence of interest. Sequences with probabilities
for a window of 10 sequences around the sequence of interest. Sequences with probabilities  or
or  are not shown. The indices (0), (1), (2), etc mark the locations of sequences observed zero, once, twice (etc) in the Lee database. Each dot corresponds to a unique sequence.
are not shown. The indices (0), (1), (2), etc mark the locations of sequences observed zero, once, twice (etc) in the Lee database. Each dot corresponds to a unique sequence.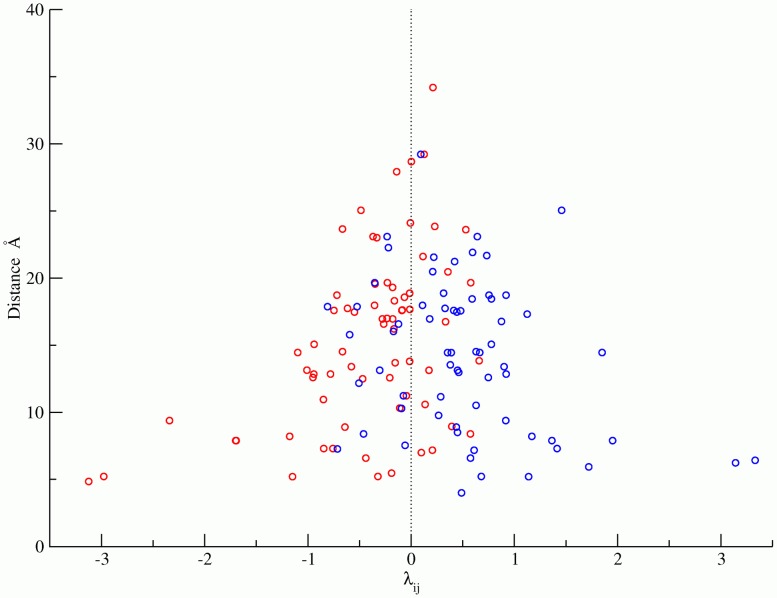
 is a fitting parameter that describes the statistical interaction energy between pairs of states. Since the Bethe mean field pair correlation model is a good approximation for this data, a negative
is a fitting parameter that describes the statistical interaction energy between pairs of states. Since the Bethe mean field pair correlation model is a good approximation for this data, a negative  indicates that a pair of states is enhanced (positively correlated), while a positive
indicates that a pair of states is enhanced (positively correlated), while a positive  indicates that a pair of states is suppressed (negatively correlated). Using simple electrostatics, we observe that like-charge patterns (blue) are mostly suppressed while unlike-charge patterns (red) are enhanced. The sign of
indicates that a pair of states is suppressed (negatively correlated). Using simple electrostatics, we observe that like-charge patterns (blue) are mostly suppressed while unlike-charge patterns (red) are enhanced. The sign of  is able to correctly predict the charge patterns for
is able to correctly predict the charge patterns for  of the top 35 most significantly correlated charge pairs out of a total of 135 pairs. The p-value for the statistical significance of this result is
of the top 35 most significantly correlated charge pairs out of a total of 135 pairs. The p-value for the statistical significance of this result is  . The reason there are 135 pairs is as follows: For each pair of residues, there are 4 possible sets of like and unlike charge combinations, resulting in a total of 612 like/unlike charge pair combinations. However, not all pairs exist in the database or are significantly correlated. Filtering results in 135 pairs with probability greater than 0.001%.
. The reason there are 135 pairs is as follows: For each pair of residues, there are 4 possible sets of like and unlike charge combinations, resulting in a total of 612 like/unlike charge pair combinations. However, not all pairs exist in the database or are significantly correlated. Filtering results in 135 pairs with probability greater than 0.001%.Similar articles
-
Accessory mutations balance the marginal stability of the HIV-1 protease in drug resistance.Proteins. 2020 Mar;88(3):476-484. doi: 10.1002/prot.25826. Epub 2019 Oct 21. Proteins. 2020. PMID: 31599014
-
A major role for a set of non-active site mutations in the development of HIV-1 protease drug resistance.Biochemistry. 2003 Jan 28;42(3):631-8. doi: 10.1021/bi027019u. Biochemistry. 2003. PMID: 12534275
-
Combining mutations in HIV-1 protease to understand mechanisms of resistance.Proteins. 2002 Jul 1;48(1):107-16. doi: 10.1002/prot.10140. Proteins. 2002. PMID: 12012342
-
HIV protease: enzyme function and drug resistance.Vitam Horm. 2000;58:213-56. doi: 10.1016/s0083-6729(00)58026-1. Vitam Horm. 2000. PMID: 10668400 Review.
-
Structural and thermodynamic basis of resistance to HIV-1 protease inhibition: implications for inhibitor design.Curr Drug Targets Infect Disord. 2003 Dec;3(4):311-28. doi: 10.2174/1568005033481051. Curr Drug Targets Infect Disord. 2003. PMID: 14754432 Review.
Cited by
-
Constructing sequence-dependent protein models using coevolutionary information.Protein Sci. 2016 Jan;25(1):111-22. doi: 10.1002/pro.2758. Epub 2015 Aug 10. Protein Sci. 2016. PMID: 26223372 Free PMC article.
-
First Passage Times, Lifetimes, and Relaxation Times of Unfolded Proteins.Phys Rev Lett. 2015 Jul 24;115(4):048101. doi: 10.1103/PhysRevLett.115.048101. Epub 2015 Jul 21. Phys Rev Lett. 2015. PMID: 26252709 Free PMC article.
-
Deep sequencing of protease inhibitor resistant HIV patient isolates reveals patterns of correlated mutations in Gag and protease.PLoS Comput Biol. 2015 Apr 20;11(4):e1004249. doi: 10.1371/journal.pcbi.1004249. eCollection 2015 Apr. PLoS Comput Biol. 2015. PMID: 25894830 Free PMC article.
-
Toward rationally redesigning bacterial two-component signaling systems using coevolutionary information.Proc Natl Acad Sci U S A. 2014 Feb 4;111(5):E563-71. doi: 10.1073/pnas.1323734111. Epub 2014 Jan 21. Proc Natl Acad Sci U S A. 2014. PMID: 24449878 Free PMC article.
-
Kinetic coevolutionary models predict the temporal emergence of HIV-1 resistance mutations under drug selection pressure.Proc Natl Acad Sci U S A. 2024 Apr 9;121(15):e2316662121. doi: 10.1073/pnas.2316662121. Epub 2024 Apr 1. Proc Natl Acad Sci U S A. 2024. PMID: 38557187 Free PMC article.
References
-
- Depristo MA, Weinreich DM, Hartl DL (2005) Missense meanderings in sequence space: A bio-physical view of protein evolution. Nature 6: 678–687. - PubMed
-
- Pace CN (1975) The stability of globular proteins. CRC Crit Rev Biochem 3: 1–43. - PubMed
-
- Pain R (1987) Temperature and macromolecular structure and function. Symp Soc Exp Biol 41: 21–33. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
