

Glucagon secretion and signaling in the development of diabetes
- PMID: 22969729
- PMCID: PMC3432929
- DOI: 10.3389/fphys.2012.00349
Glucagon secretion and signaling in the development of diabetes
Abstract
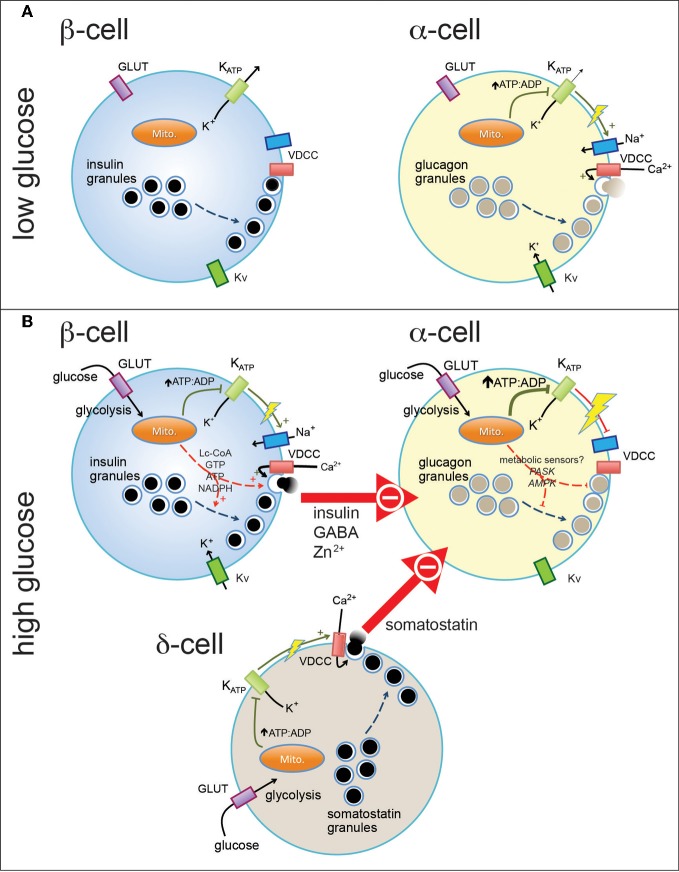
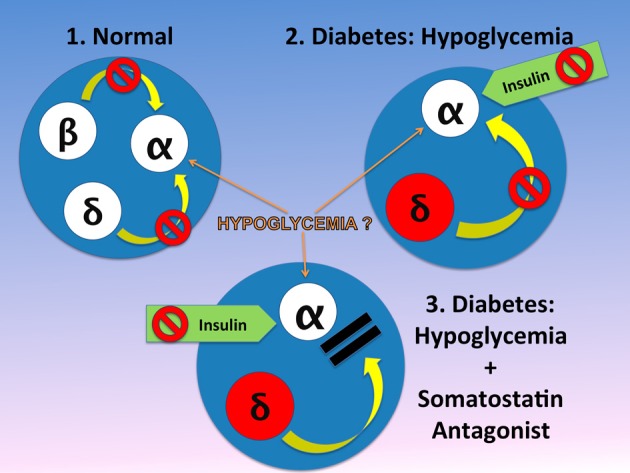
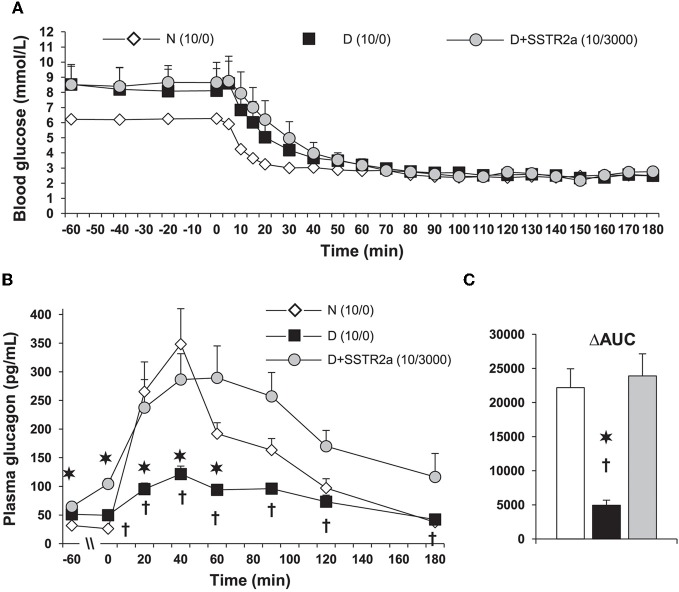
Normal release of glucagon from pancreatic islet α-cells promotes glucose mobilization, which counteracts the hypoglycemic actions of insulin, thereby ensuring glucose homeostasis. In treatment of diabetes aimed at rigorously reducing hyperglycemia to avoid chronic complications, the resulting hypoglycemia triggering glucagon release from α-cells is frequently impaired, with ensuing hypoglycemic complications. This review integrates the physiology of glucagon secretion regulating glucose homeostasis in vivo to single α-cell signaling, and how both become perturbed in diabetes. α-cells within the social milieu of the islet micro-organ are regulated not only by intrinsic signaling events but also by paracrine regulation, particularly by adjacent insulin-secreting β-cells and somatostatin-secreting δ-cells. We discuss the intrinsic α-cell signaling events, including glucose sensing and ion channel regulation leading to glucagon secretion. We then discuss the complex crosstalk between the islet cells and the breakdown of this crosstalk in diabetes contributing to the dysregulated glucagon secretion. Whereas, there are many secretory products released by β- and δ-cells that become deficient or excess in diabetes, we discuss the major ones, including the better known insulin and lesser known somatostatin, which act as putative paracrine on/off switches that very finely regulate α-cell secretory responses in health and diabetes. Of note in several type 1 diabetes (T1D) rodent models, blockade of excess somatostatin actions on α-cell could normalize glucagon secretion sufficient to attain normoglycemia in response to hypoglycemic assaults. There has been slow progress in fully elucidating the pathophysiology of the α-cell in diabetes because of the small number of α-cells within an islet and the islet mass becomes severely reduced and inflamed in diabetes. These limitations are just now being surmounted by new approaches.
Keywords: diabetes; glucagon secretion; hypoglycemia; islet α-cell; somatostatin.
Figures
References
-
- Amatruda J., Livingston J. (2003). Glucagon, in, Ellenberg and Rifkin's Diabetes Mellitus, eds Porte D., Jr., Sherwin R., Baron A. (New York, NY: McGraw-Hill; ), 97–115
LinkOut - more resources
Full Text Sources