

Autoimmune dysregulation and purine metabolism in adenosine deaminase deficiency
- PMID: 22969765
- PMCID: PMC3427915
- DOI: 10.3389/fimmu.2012.00265
Autoimmune dysregulation and purine metabolism in adenosine deaminase deficiency
Abstract
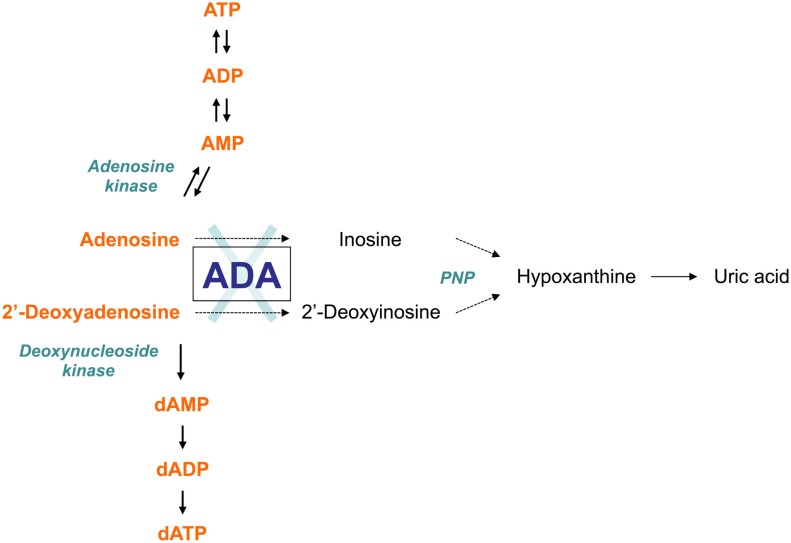
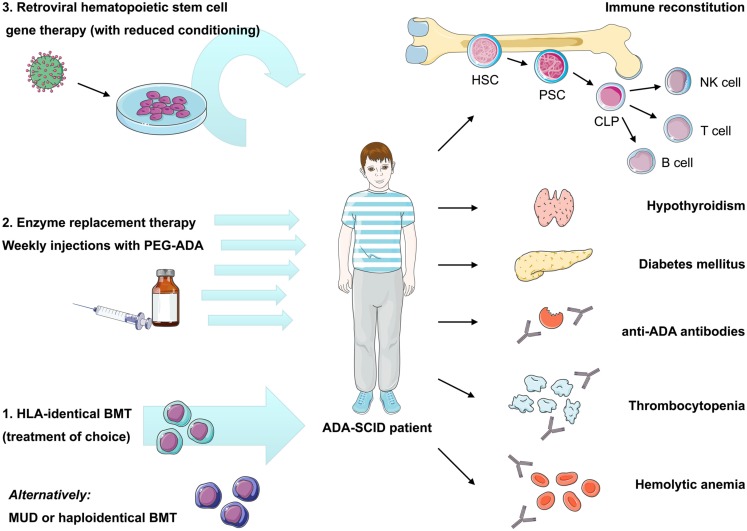
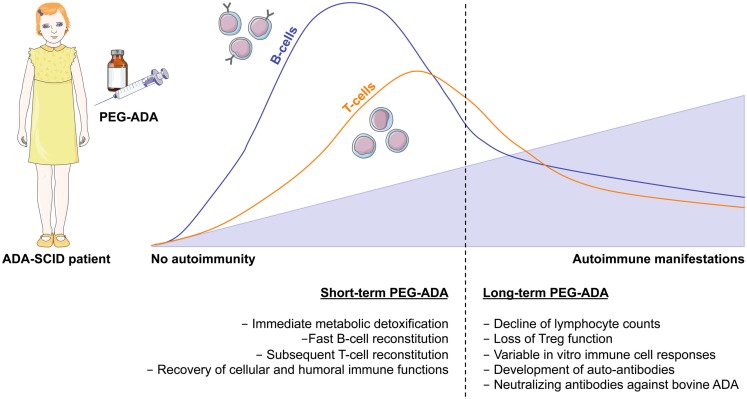
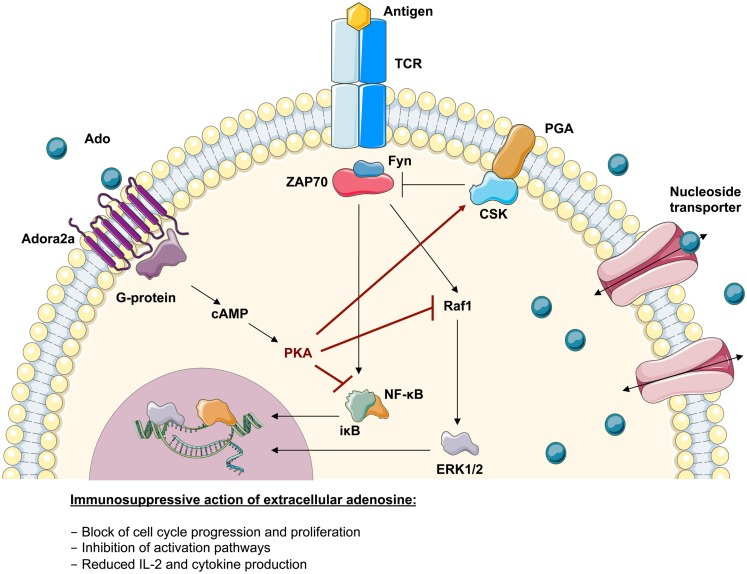
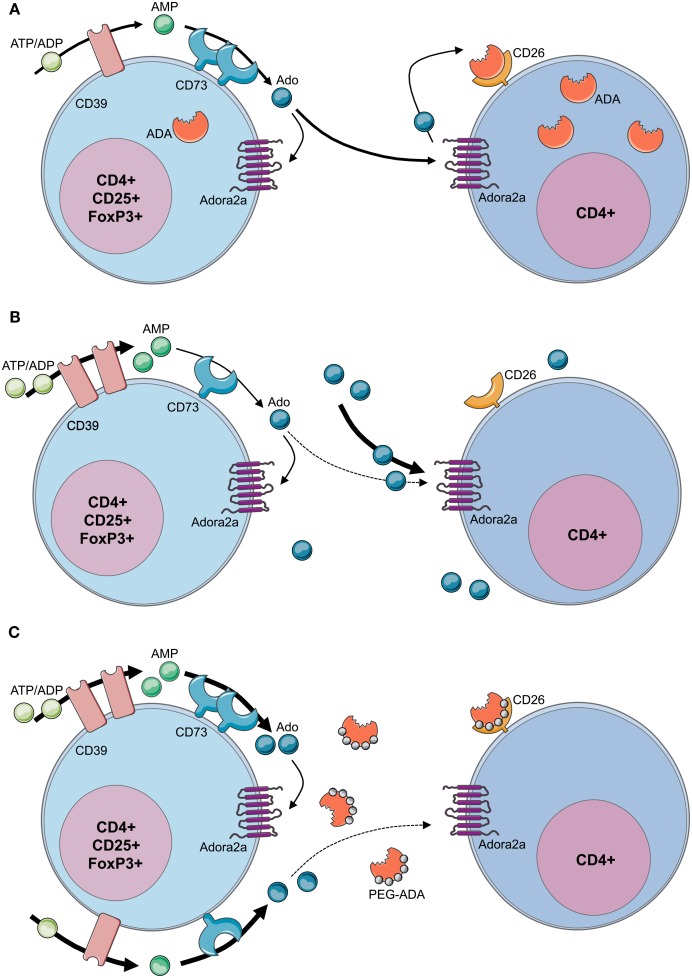
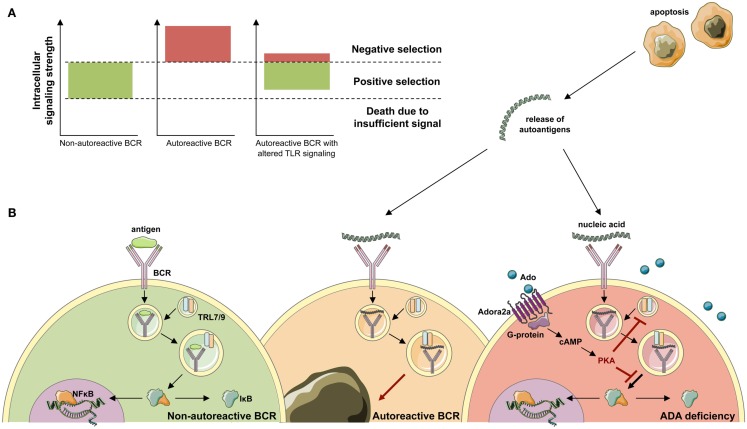
Genetic defects in the adenosine deaminase (ADA) gene are among the most common causes for severe combined immunodeficiency (SCID). ADA-SCID patients suffer from lymphopenia, severely impaired cellular and humoral immunity, failure to thrive, and recurrent infections. Currently available therapeutic options for this otherwise fatal disorder include bone marrow transplantation (BMT), enzyme replacement therapy with bovine ADA (PEG-ADA), or hematopoietic stem cell gene therapy (HSC-GT). Although varying degrees of immune reconstitution can be achieved by these treatments, breakdown of tolerance is a major concern in ADA-SCID. Immune dysregulation such as autoimmune hypothyroidism, diabetes mellitus, hemolytic anemia, and immune thrombocytopenia are frequently observed in milder forms of the disease. However, several reports document similar complications also in patients on long-term PEG-ADA and after BMT or GT treatment. A skewed repertoire and decreased immune functions have been implicated in autoimmunity observed in certain B-cell and/or T-cell immunodeficiencies, but it remains unclear to what extent specific mechanisms of tolerance are affected in ADA deficiency. Herein we provide an overview about ADA-SCID and the autoimmune manifestations reported in these patients before and after treatment. We also assess the value of the ADA-deficient mouse model as a useful tool to study both immune and metabolic disease mechanisms. With focus on regulatory T- and B-cells we discuss the lymphocyte subpopulations particularly prone to contribute to the loss of self-tolerance and onset of autoimmunity in ADA deficiency. Moreover we address which aspects of immune dysregulation are specifically related to alterations in purine metabolism caused by the lack of ADA and the subsequent accumulation of metabolites with immunomodulatory properties.
Keywords: ADA-SCID; adenosine deaminase; autoimmunity; gene therapy; severe combined immunodeficiency.
Figures
References
-
- Abuchowski A., van Es T., Palczuk N. C., Davis F. F. (1977). Alteration of immunological properties of bovine serum albumin by covalent attachment of polyethylene glycol. J. Biol. Chem. 252, 3578–3581 - PubMed
-
- Aiuti A., Cassani B., Andolfi G., Mirolo M., Biasco L., Recchia A., Urbinati F., Valacca C., Scaramuzza S., Aker M., Slavin S., Cazzola M., Sartori D., Ambrosi A., Di Serio C., Roncarolo M. G., Mavilio F., Bordignon C. (2007). Multilineage hematopoietic reconstitution without clonal selection in ADA-SCID patients treated with stem cell gene therapy. J. Clin. Invest. 117, 2233–2240 10.1172/JCI31666 - DOI - PMC - PubMed
-
- Aiuti A., Cattaneo F., Galimberti F., Benninghoff U., Cassani B., Callegaro L., Scaramuzza S., Andolfi G., Mirolo M., Brigida I., Tabucchi A., Carlucci F., Eibl M., Aker M., Slavin S., Al-Mousa H., Al Ghonaium A., Ferster A., Duppenthaler A., Notarangelo L., Wintergerst U., Buckley R., Bregni M., Valsecchi M., Rossi P., Ciceri F., Miniero R., Bordignon C., Roncarolo M. (2009). Long-term safety and efficacy of gene therapy for adenosine deaminase (ADA)-deficient severe combined immunodeficiency. N. Engl. J. Med. 360, 447–458 10.1056/NEJMoa0805817 - DOI - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous