

Technical reproducibility of genotyping SNP arrays used in genome-wide association studies
- PMID: 22970228
- PMCID: PMC3436888
- DOI: 10.1371/journal.pone.0044483
Technical reproducibility of genotyping SNP arrays used in genome-wide association studies
Abstract
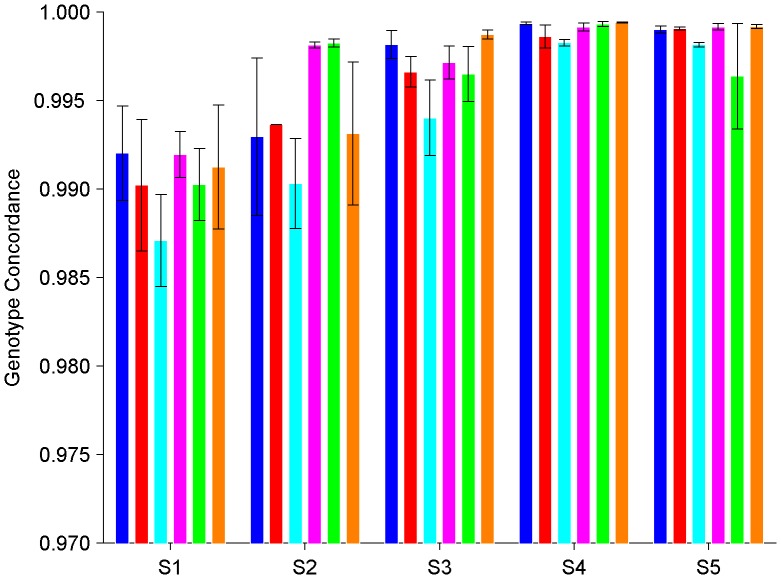
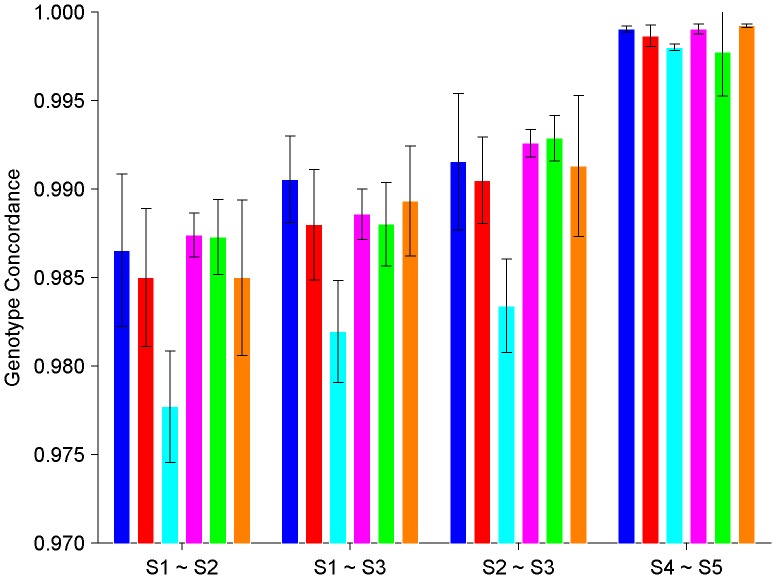
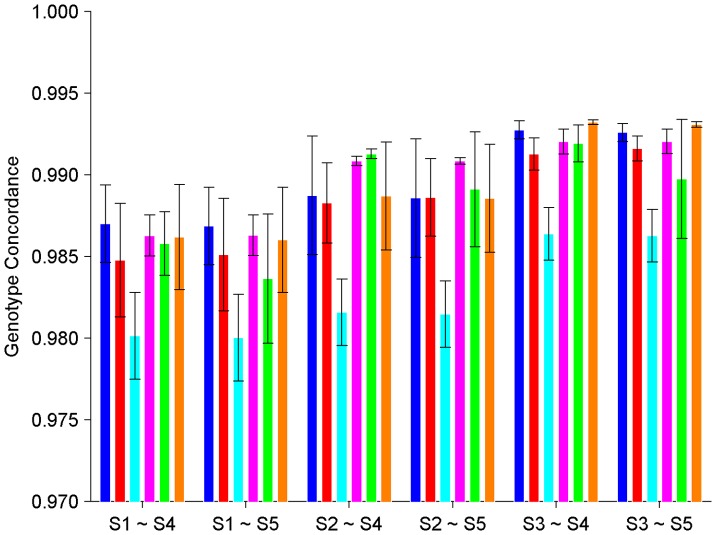
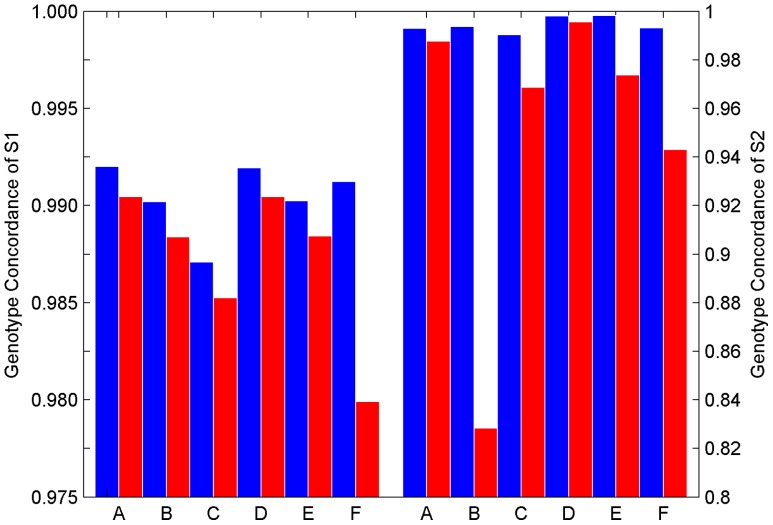
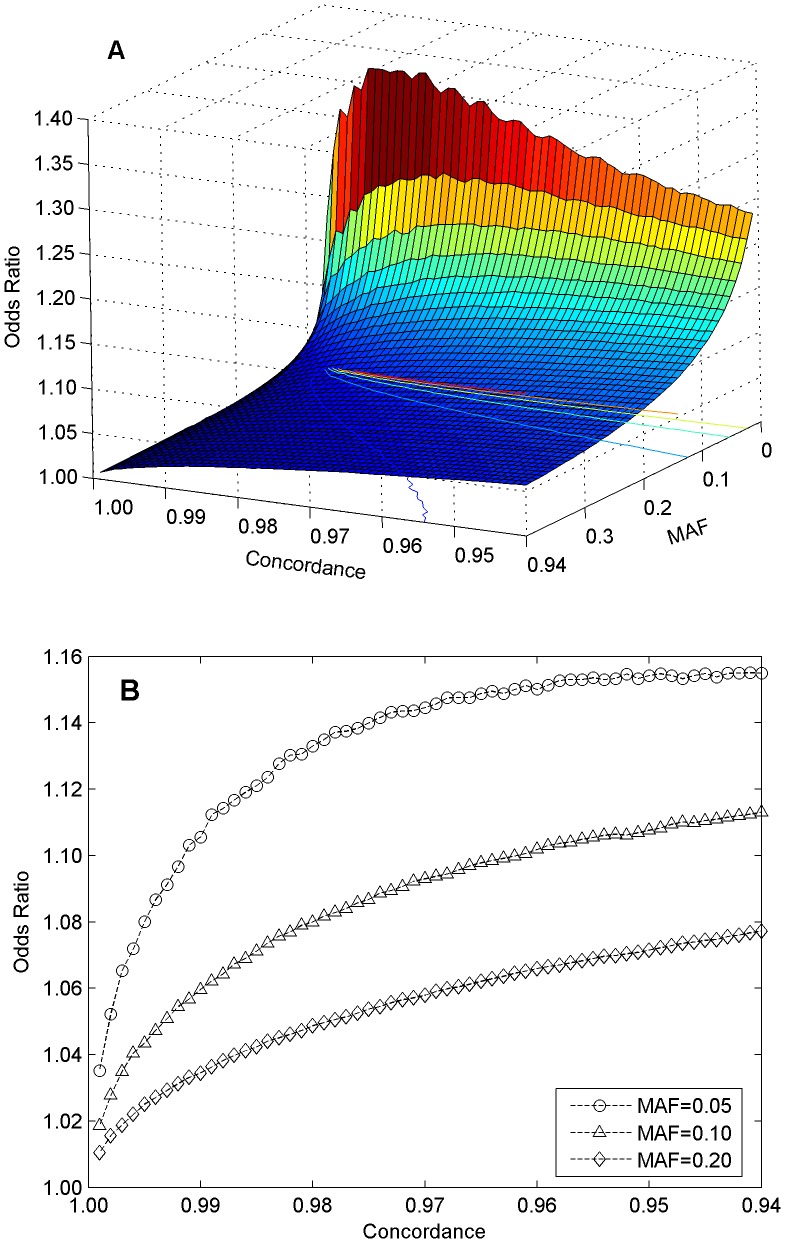
During the last several years, high-density genotyping SNP arrays have facilitated genome-wide association studies (GWAS) that successfully identified common genetic variants associated with a variety of phenotypes. However, each of the identified genetic variants only explains a very small fraction of the underlying genetic contribution to the studied phenotypic trait. Moreover, discordance observed in results between independent GWAS indicates the potential for Type I and II errors. High reliability of genotyping technology is needed to have confidence in using SNP data and interpreting GWAS results. Therefore, reproducibility of two widely genotyping technology platforms from Affymetrix and Illumina was assessed by analyzing four technical replicates from each of the six individuals in five laboratories. Genotype concordance of 99.40% to 99.87% within a laboratory for the sample platform, 98.59% to 99.86% across laboratories for the same platform, and 98.80% across genotyping platforms was observed. Moreover, arrays with low quality data were detected when comparing genotyping data from technical replicates, but they could not be detected according to venders' quality control (QC) suggestions. Our results demonstrated the technical reliability of currently available genotyping platforms but also indicated the importance of incorporating some technical replicates for genotyping QC in order to improve the reliability of GWAS results. The impact of discordant genotypes on association analysis results was simulated and could explain, at least in part, the irreproducibility of some GWAS findings when the effect size (i.e. the odds ratio) and the minor allele frequencies are low.
Conflict of interest statement
Figures
References
-
- Sladek R, Rocheleau G, Rung J, Dina C, Shen L, et al. (2007) A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature 445: 881–885. - PubMed
MeSH terms
LinkOut - more resources
Full Text Sources
Research Materials
