

Transforming growth factor-beta promotes rhinovirus replication in bronchial epithelial cells by suppressing the innate immune response
- PMID: 22970254
- PMCID: PMC3435262
- DOI: 10.1371/journal.pone.0044580
Transforming growth factor-beta promotes rhinovirus replication in bronchial epithelial cells by suppressing the innate immune response
Abstract
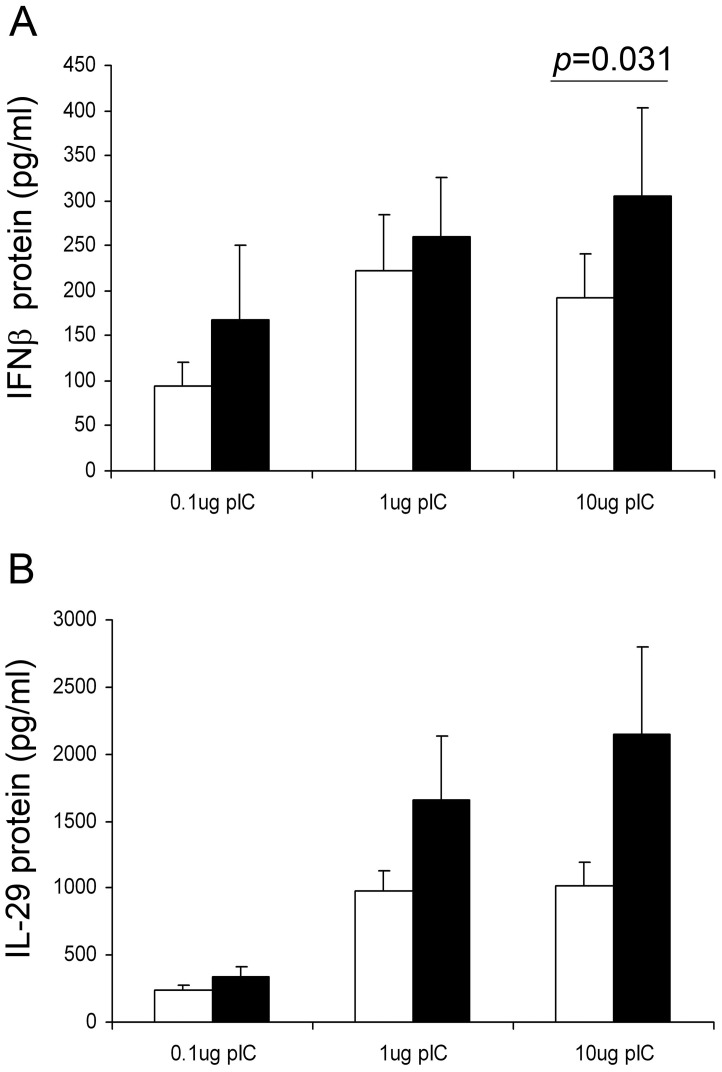
Rhinovirus (RV) infection is a major cause of asthma exacerbations which may be due to a deficient innate immune response in the bronchial epithelium. We hypothesized that the pleiotropic cytokine, TGF-β, influences interferon (IFN) production by primary bronchial epithelial cells (PBECs) following RV infection. Exogenous TGF-β(2) increased RV replication and decreased IFN protein secretion in response to RV or double-stranded RNA (dsRNA). Conversely, neutralizing TGF-β antibodies decreased RV replication and increased IFN expression in response to RV or dsRNA. Endogenous TGF-β(2) levels were higher in conditioned media of PBECs from asthmatic donors and the suppressive effect of anti-TGF-β on RV replication was significantly greater in these cells. Basal SMAD-2 activation was reduced when asthmatic PBECs were treated with anti-TGF-β and this was accompanied by suppression of SOCS-1 and SOCS-3 expression. Our results suggest that endogenous TGF-β contributes to a suppressed IFN response to RV infection possibly via SOCS-1 and SOCS-3.
Conflict of interest statement
Figures








References
-
- Holgate ST, Polosa R (2006) The mechanisms, diagnosis, and management of severe asthma in adults. Lancet 368: 780–793. - PubMed
-
- Cohn L, Elias JA, Chupp GL (2004) Asthma: mechanisms of disease persistence and progression. Annu Rev Immunol 22: 789–815. - PubMed
-
- Contoli M, Message SD, Laza-Stanca V, Edwards MR, Wark PA, et al. (2006) Role of deficient type III interferon-lambda production in asthma exacerbations. Nat Med 12: 1023–1026. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
