

Whole genome sequencing of field isolates provides robust characterization of genetic diversity in Plasmodium vivax
- PMID: 22970335
- PMCID: PMC3435244
- DOI: 10.1371/journal.pntd.0001811
Whole genome sequencing of field isolates provides robust characterization of genetic diversity in Plasmodium vivax
Abstract
Background: An estimated 2.85 billion people live at risk of Plasmodium vivax transmission. In endemic countries vivax malaria causes significant morbidity and its mortality is becoming more widely appreciated, drug-resistant strains are increasing in prevalence, and an increasing number of reports indicate that P. vivax is capable of breaking through the Duffy-negative barrier long considered to confer resistance to blood stage infection. Absence of robust in vitro propagation limits our understanding of fundamental aspects of the parasite's biology, including the determinants of its dormant hypnozoite phase, its virulence and drug susceptibility, and the molecular mechanisms underlying red blood cell invasion.
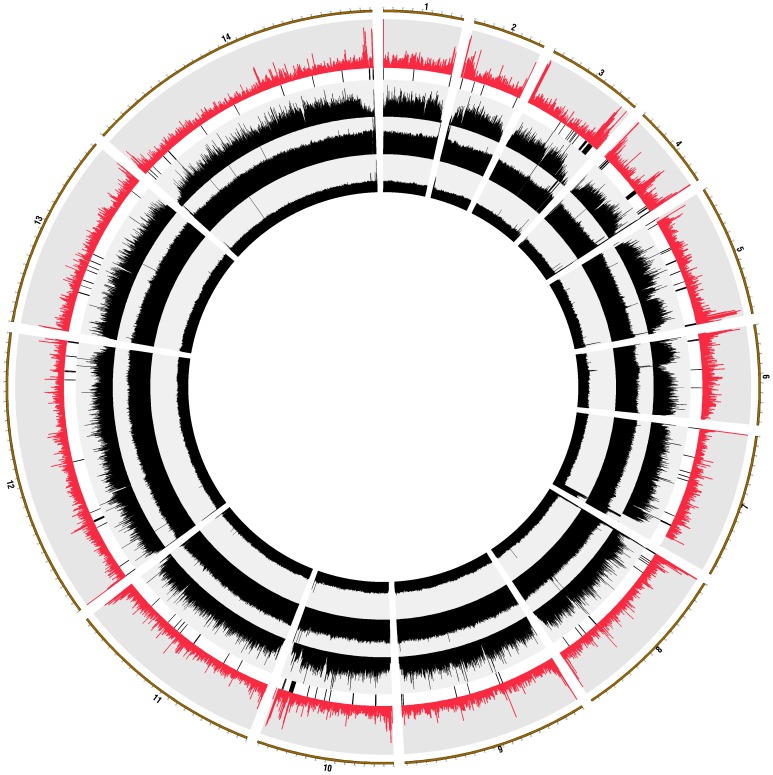
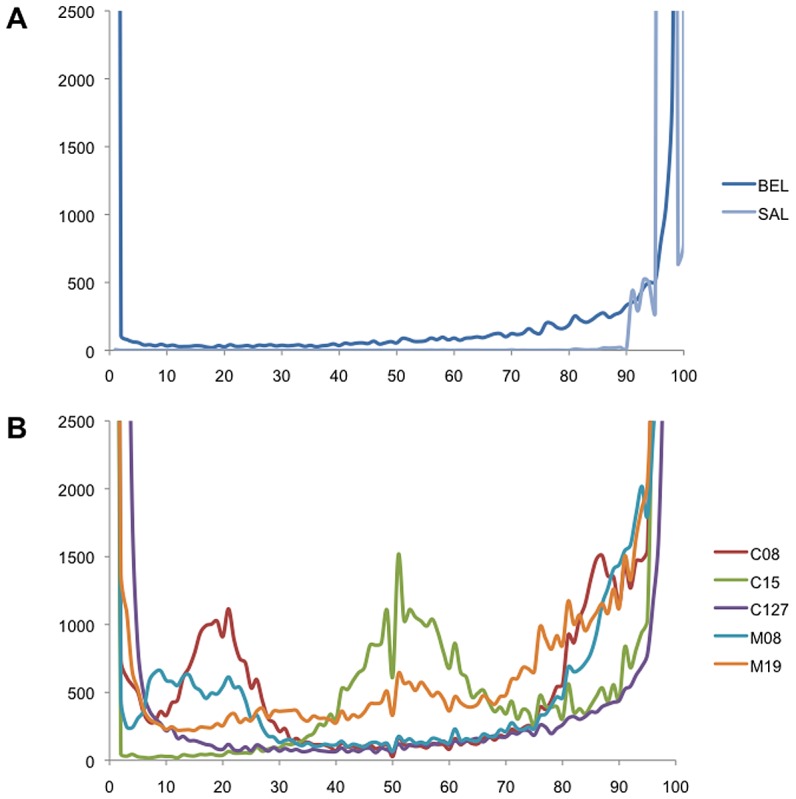
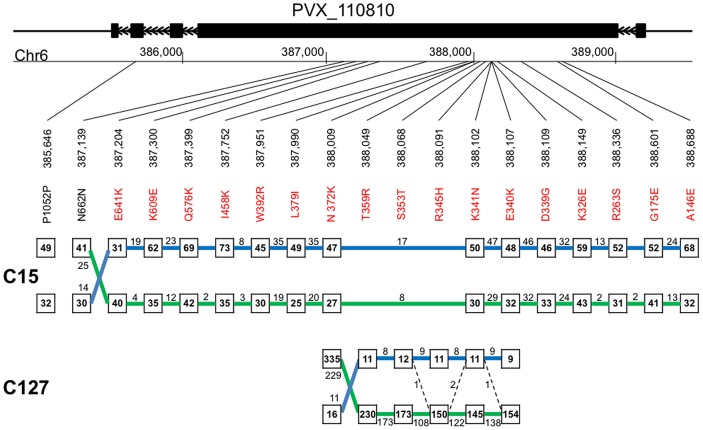
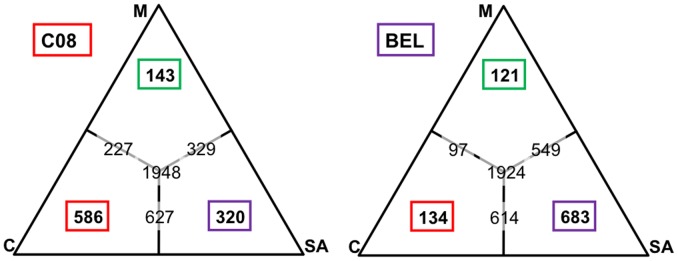
Methodology/principal findings: Here, we report results from whole genome sequencing of five P. vivax isolates obtained from Malagasy and Cambodian patients, and of the monkey-adapted Belem strain. We obtained an average 70-400 X coverage of each genome, resulting in more than 93% of the Sal I reference sequence covered by 20 reads or more. Our study identifies more than 80,000 SNPs distributed throughout the genome which will allow designing association studies and population surveys. Analysis of the genome-wide genetic diversity in P. vivax also reveals considerable allele sharing among isolates from different continents. This observation could be consistent with a high level of gene flow among parasite strains distributed throughout the world.
Conclusions: Our study shows that it is feasible to perform whole genome sequencing of P. vivax field isolates and rigorously characterize the genetic diversity of this parasite. The catalogue of polymorphisms generated here will enable large-scale genotyping studies and contribute to a better understanding of P. vivax traits such as drug resistance or erythrocyte invasion, partially circumventing the lack of laboratory culture that has hampered vivax research for years.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- Mendis K, Sina BJ, Marchesini P, Carter R (2001) The neglected burden of Plasmodium vivax malaria. Am J Trop Med Hyg 64: 97–106. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
