

Phenotypic heterogeneity of genomic disorders and rare copy-number variants
- PMID: 22970919
- PMCID: PMC3494411
- DOI: 10.1056/NEJMoa1200395
Phenotypic heterogeneity of genomic disorders and rare copy-number variants
Erratum in
- N Engl J Med. 2012 Dec 13;367(24):2362
Abstract
Background: Some copy-number variants are associated with genomic disorders with extreme phenotypic heterogeneity. The cause of this variation is unknown, which presents challenges in genetic diagnosis, counseling, and management.
Methods: We analyzed the genomes of 2312 children known to carry a copy-number variant associated with intellectual disability and congenital abnormalities, using array comparative genomic hybridization.
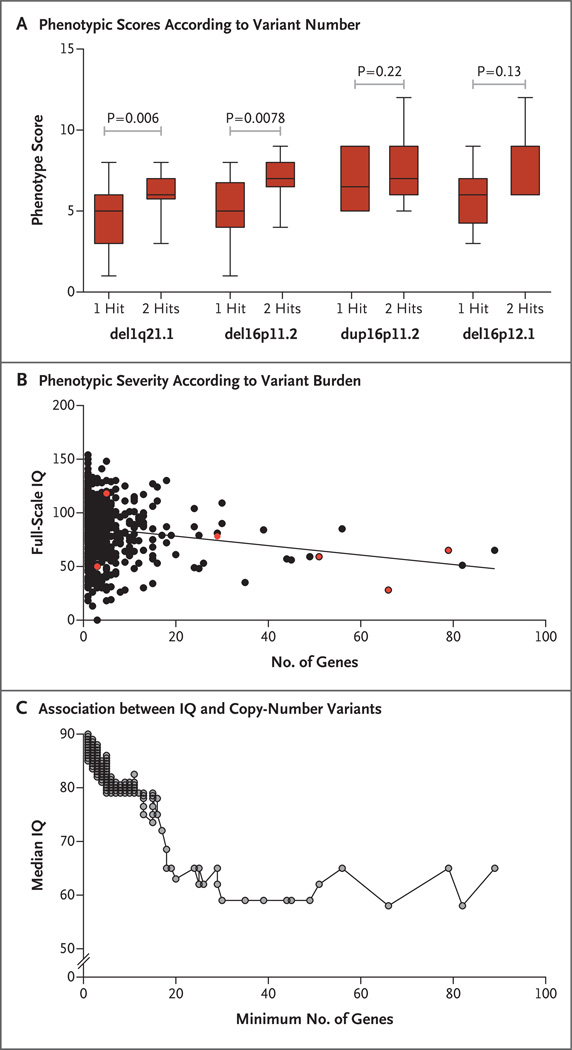
Results: Among the affected children, 10.1% carried a second large copy-number variant in addition to the primary genetic lesion. We identified seven genomic disorders, each defined by a specific copy-number variant, in which the affected children were more likely to carry multiple copy-number variants than were controls. We found that syndromic disorders could be distinguished from those with extreme phenotypic heterogeneity on the basis of the total number of copy-number variants and whether the variants are inherited or de novo. Children who carried two large copy-number variants of unknown clinical significance were eight times as likely to have developmental delay as were controls (odds ratio, 8.16; 95% confidence interval, 5.33 to 13.07; P=2.11×10(-38)). Among affected children, inherited copy-number variants tended to co-occur with a second-site large copy-number variant (Spearman correlation coefficient, 0.66; P<0.001). Boys were more likely than girls to have disorders of phenotypic heterogeneity (P<0.001), and mothers were more likely than fathers to transmit second-site copy-number variants to their offspring (P=0.02).
Conclusions: Multiple, large copy-number variants, including those of unknown pathogenic significance, compound to result in a severe clinical presentation, and secondary copy-number variants are preferentially transmitted from maternal carriers. (Funded by the Simons Foundation Autism Research Initiative and the National Institutes of Health.).
Figures


Comment in
-
The variability of genetic disease.N Engl J Med. 2012 Oct 4;367(14):1350-2. doi: 10.1056/NEJMe1209699. Epub 2012 Sep 12. N Engl J Med. 2012. PMID: 22970918 No abstract available.
References
-
- Antonell A, Del Campo M, Magano LF, et al. Partial 7q11.23 deletions further implicate GTF2I and GTF2IRD1 as the main genes responsible for the Williams-Beuren syndrome neurocognitive profile. J Med Genet. 2010;47:312–320. - PubMed
-
- Girirajan S, Vlangos CN, Szomju BB, et al. Genotype-phenotype correlation in Smith-Magenis syndrome: evidence that multiple genes in 17p11.2 contribute to the clinical spectrum. Genet Med. 2006;8:417–427. - PubMed
-
- Liburd N, Ghosh M, Riazuddin S, et al. Novel mutations of MYO15A associated with profound deafness in consanguineous families and moderately severe hearing loss in a patient with Smith-Magenis syndrome. Hum Genet. 2001;109:535–541. - PubMed
-
- Sarasua SM, Dwivedi A, Boccuto L, et al. Association between deletion size and important phenotypes expands the genomic region of interest in Phelan-McDermid syndrome (22q13 deletion syndrome) J Med Genet. 2011;48:761–766. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases