

Implementation of a reference-scaled average bioequivalence approach for highly variable generic drug products by the US Food and Drug Administration
- PMID: 22972221
- PMCID: PMC3475857
- DOI: 10.1208/s12248-012-9406-x
Implementation of a reference-scaled average bioequivalence approach for highly variable generic drug products by the US Food and Drug Administration
Abstract
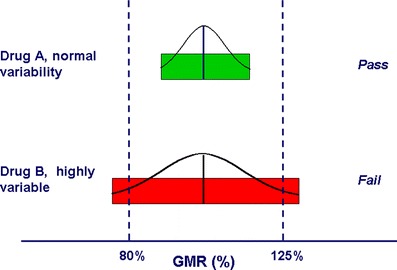
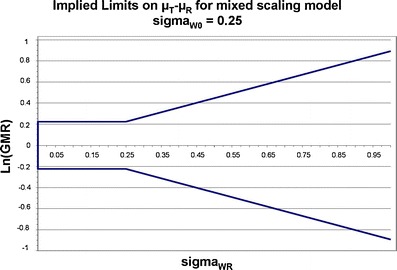
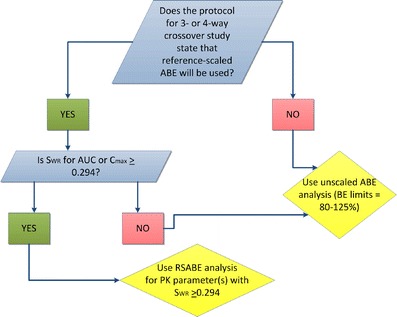
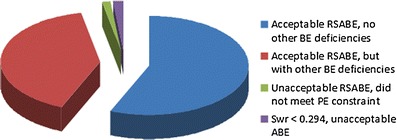
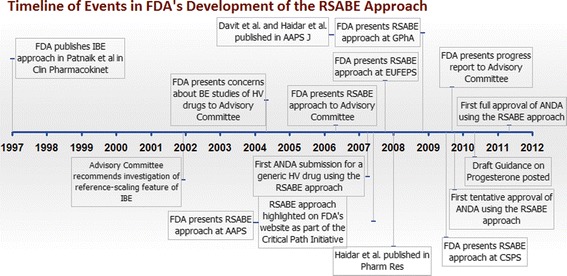
Highly variable (HV) drugs are defined as those for which within-subject variability (%CV) in bioequivalence (BE) measures is 30% or greater. Because of this high variability, studies designed to show whether generic HV drugs are bioequivalent to their corresponding HV reference drugs may need to enroll large numbers of subjects even when the products have no significant mean differences. To avoid unnecessary human testing, the US Food and Drug Administration's Office of Generic Drugs developed a reference-scaled average bioequivalence (RSABE) approach, whereby the BE acceptance limits are scaled to the variability of the reference product. For an acceptable RSABE study, an HV generic drug product must meet the scaled BE limit and a point estimate constraint. The approach has been implemented successfully. To date, the RSABE approach has supported four full approvals and one tentative approval of HV generic drug products.
Figures
References
-
- Federal Food, Drug, and Cosmetic Act, Chapter V, Subchapter A, Drugs and Devices Section 355(j)(8)(B)(i).
-
- Schuirmann DJ. A comparison of the two one-sided tests procedure and the power approach for assessing the equivalence of average bioavailability. J Pharmacokinet Biopharm. 1987;15:657–80. - PubMed
-
- Westlake WJ. Bioequivalence testing: a need to rethink. Biometrics. 1981;37:589–94. doi: 10.2307/2530573. - DOI
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
