

Challenges and opportunities in estimating viral genetic diversity from next-generation sequencing data
- PMID: 22973268
- PMCID: PMC3438994
- DOI: 10.3389/fmicb.2012.00329
Challenges and opportunities in estimating viral genetic diversity from next-generation sequencing data
Abstract
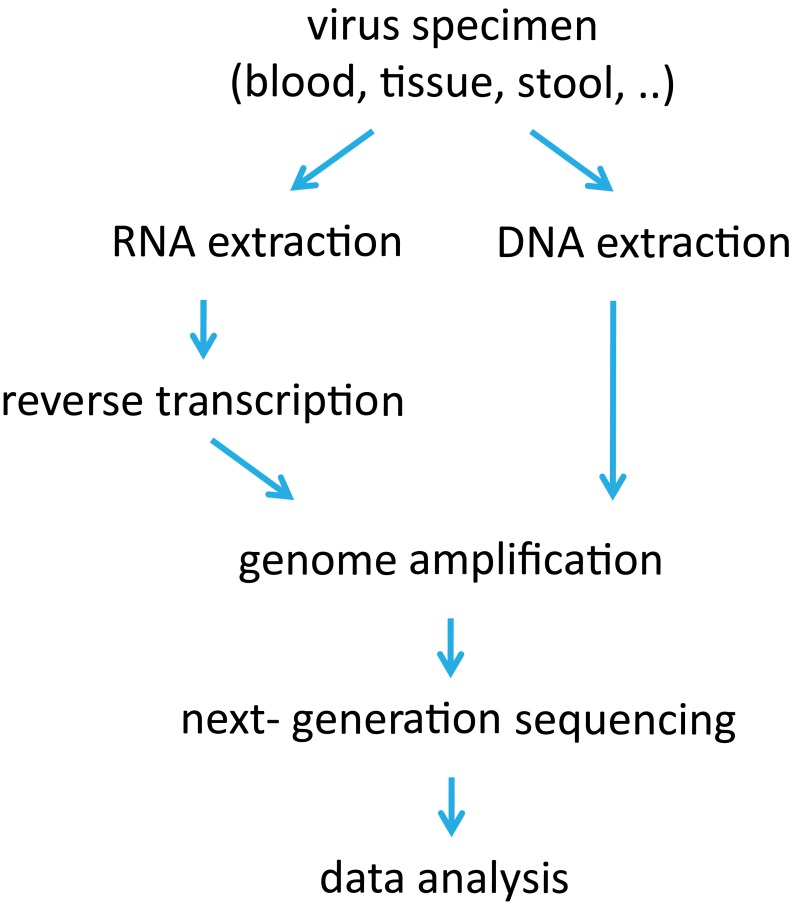
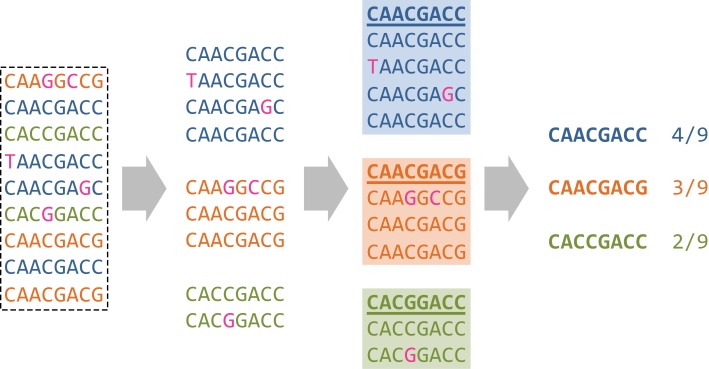
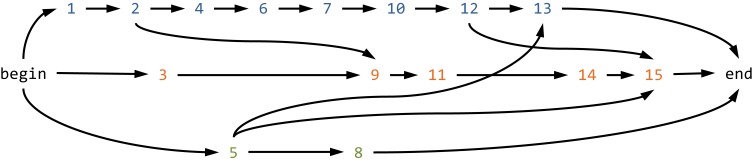
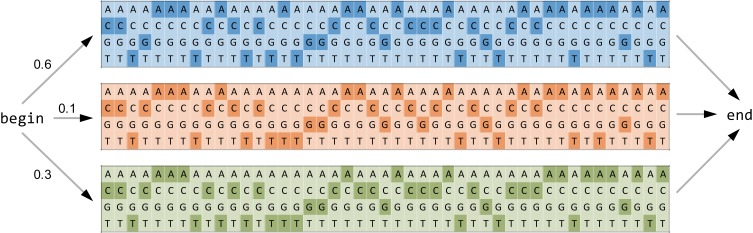
Many viruses, including the clinically relevant RNA viruses HIV (human immunodeficiency virus) and HCV (hepatitis C virus), exist in large populations and display high genetic heterogeneity within and between infected hosts. Assessing intra-patient viral genetic diversity is essential for understanding the evolutionary dynamics of viruses, for designing effective vaccines, and for the success of antiviral therapy. Next-generation sequencing (NGS) technologies allow the rapid and cost-effective acquisition of thousands to millions of short DNA sequences from a single sample. However, this approach entails several challenges in experimental design and computational data analysis. Here, we review the entire process of inferring viral diversity from sample collection to computing measures of genetic diversity. We discuss sample preparation, including reverse transcription and amplification, and the effect of experimental conditions on diversity estimates due to in vitro base substitutions, insertions, deletions, and recombination. The use of different NGS platforms and their sequencing error profiles are compared in the context of various applications of diversity estimation, ranging from the detection of single nucleotide variants (SNVs) to the reconstruction of whole-genome haplotypes. We describe the statistical and computational challenges arising from these technical artifacts, and we review existing approaches, including available software, for their solution. Finally, we discuss open problems, and highlight successful biomedical applications and potential future clinical use of NGS to estimate viral diversity.
Keywords: bioinformatics; error correction; haplotype inference; next-generation sequencing; quasispecies assembly; statistics; viral diversity; viral quasispecies.
Figures

References
-
- Abbate I., Vlassi C., Rozera G., Bruselles A., Bartolini B., Giombini E., Corpolongo A., D'Offizi G., Narciso P., Desideri A., Ippolito G., Capobianchi M. R. (2011). Detection of quasispecies variants predicted to use CXCR4 by ultra-deep pyrosequencing during early HIV infection. AIDS 25, 611–617 10.1097/QAD.0b013e328343489e - DOI - PubMed
-
- Alteri C., Santoro M. M., Abbate I., Rozera G., Bruselles A., Bartolini B., Gori C., Forbici F., Orchi N., Tozzi V., Palamara G., Antinori A., Narciso P., Girardi E., Svicher V., Ceccherini-Silberstein F., Capobianchi M. R., Perno C. F. (2011). ‘Sentinel’ mutations in standard population sequencing can predict the presence of HIV-1 reverse transcriptase major mutations detectable only by ultra-deep pyrosequencing. J. Antimicrob. Chemother. 66, 2615–2623 10.1093/jac/dkr354 - DOI - PubMed
-
- Althaus C. F., Vongrad V., Niederost B., Joos B., Di Giallonardo F., Rieder P., Pavlovic J., Trkola A., Gunthard H. F., Metzner K. J., Fischer M. (2012). Tailored enrichment strategy detects low abundant small noncoding RNAs in HIV-1 infected cells. Retrovirology 9, 27 10.1186/1742-4690-9-27 - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources