

Rapamycin has a biphasic effect on insulin sensitivity in C2C12 myotubes due to sequential disruption of mTORC1 and mTORC2
- PMID: 22973301
- PMCID: PMC3438685
- DOI: 10.3389/fgene.2012.00177
Rapamycin has a biphasic effect on insulin sensitivity in C2C12 myotubes due to sequential disruption of mTORC1 and mTORC2
Abstract
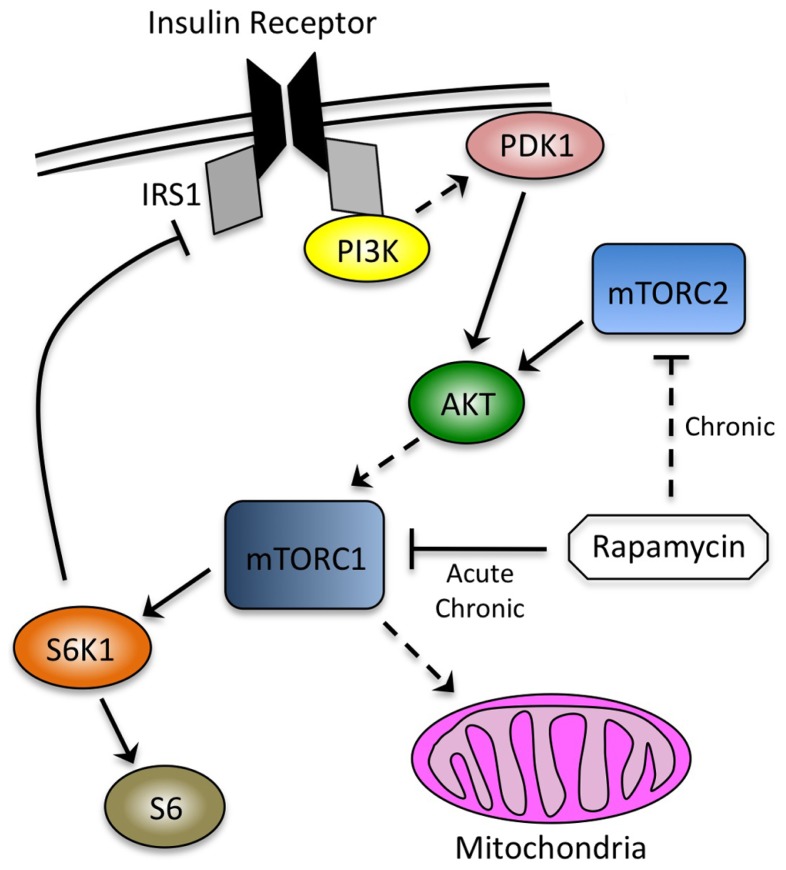
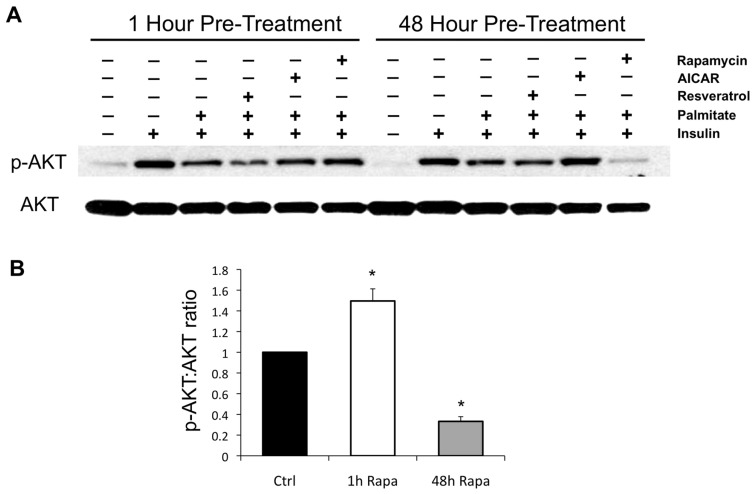
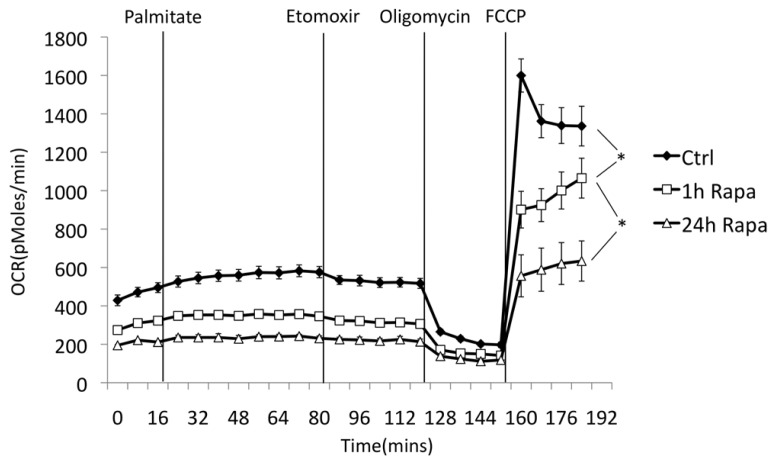
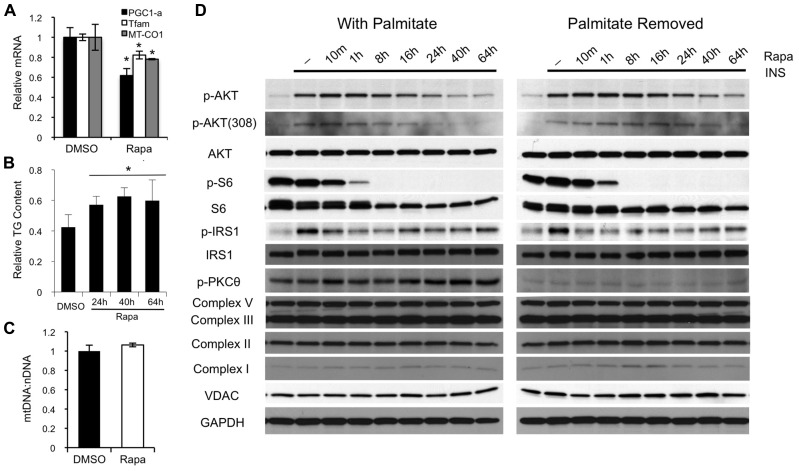
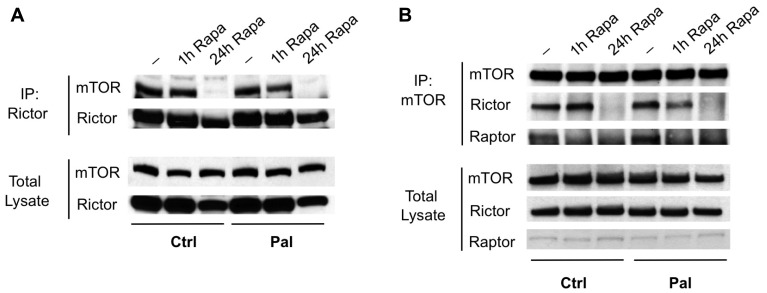
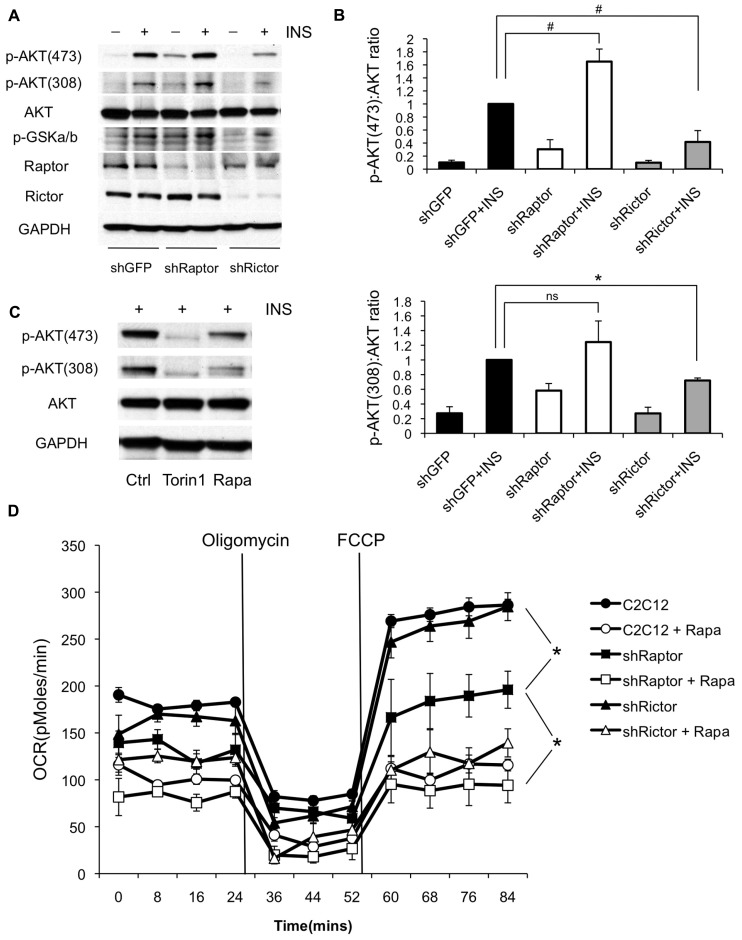
Rapamycin, an inhibitor of mTOR complex 1 (mTORC1), improves insulin sensitivity in acute studies in vitro and in vivo by disrupting a negative feedback loop mediated by S6 kinase. We find that rapamycin has a clear biphasic effect on insulin sensitivity in C2C12 myotubes, with enhanced responsiveness during the first hour that declines to almost complete insulin resistance by 24-48 h. We and others have recently observed that chronic rapamycin treatment induces insulin resistance in rodents, at least in part due to disruption of mTORC2, an mTOR-containing complex that is not acutely sensitive to the drug. Chronic rapamycin treatment may also impair insulin action via the inhibition of mTORC1-dependent mitochondrial biogenesis and activity, which could result in a buildup of lipid intermediates that are known to trigger insulin resistance. We confirmed that rapamycin inhibits expression of PGC-1α, a key mitochondrial transcription factor, and acutely reduces respiration rate in myotubes. However, rapamycin did not stimulate phosphorylation of PKCθ, a central mediator of lipid-induced insulin resistance. Instead, we found dramatic disruption of mTORC2, which coincided with the onset of insulin resistance. Selective inhibition of mTORC1 or mTORC2 by shRNA-mediated knockdown of specific components (Raptor and Rictor, respectively) confirmed that mitochondrial effects of rapamycin are mTORC1-dependent, whereas insulin resistance was recapitulated only by knockdown of mTORC2. Thus, mTORC2 disruption, rather than inhibition of mitochondria, causes insulin resistance in rapamycin-treated myotubes, and this system may serve as a useful model to understand the effects of rapamycin on mTOR signaling in vivo.
Keywords: chronic; diabetes; insulin sensitivity; mTOR; mTORC1; mTORC2; mitochondrial biogenesis; rapamycin; raptor; respiration; rictor.
Figures
References
-
- Baur J. A. (2009). “Metabolic effects of caloric restriction,” in Encyclopedia of Life Sciences (Chichester: John Wiley & Sons, Ltd).
-
- Bentzinger C. F., Romanino K., Cloetta D., Lin S., Mascarenhas J. B., Oliveri F., Xia J., Casanova E., Costa C. F., Brink M., Zorzato F., Hall M. N, Rüegg M. A. (2008). Skeletal muscle-specific ablation of raptor, but not of rictor, causes metabolic changes and results in muscle dystrophy. Cell Metab. 8 411–424 - PubMed
-
- Blattler S. M., Cunningham J. T., Verdeguer F., Chim H., Haas W., Liu H., Romanino K., Ruegg M. A., Gygi S. P., Shi Y., Puigserver P. (2012). Yin Yang 1 deficiency in skeletal muscle protects against rapamycin-induced diabetic-like symptoms through activation of insulin/IGF signaling. Cell Metab. 15 505–517 - PMC - PubMed
-
- Chavez J. A., Holland W. L., Bar J., Sandhoff K., Summers S. A. (2005). Acid ceramidase overexpression prevents the inhibitory effects of saturated fatty acids on insulin signaling. J. Biol. Chem. 280 20148–20153 - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous