

Uncovering networks from genome-wide association studies via circular genomic permutation
- PMID: 22973544
- PMCID: PMC3429921
- DOI: 10.1534/g3.112.002618
Uncovering networks from genome-wide association studies via circular genomic permutation
Abstract
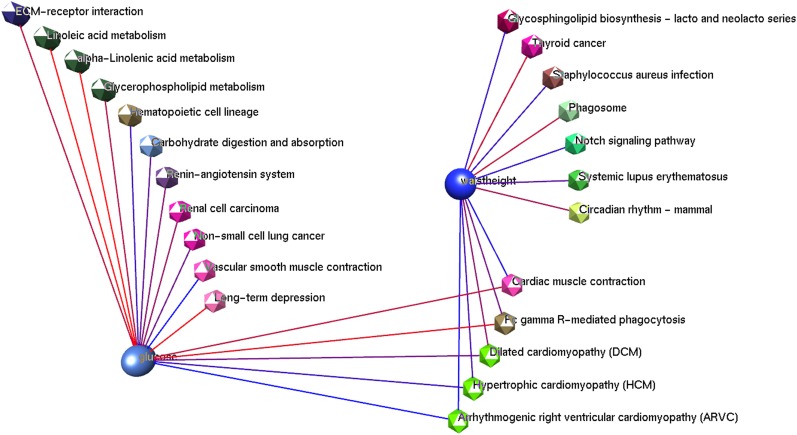
Genome-wide association studies (GWAS) aim to detect single nucleotide polymorphisms (SNP) associated with trait variation. However, due to the large number of tests, standard analysis techniques impose highly stringent significance thresholds, leaving potentially associated SNPs undetected, and much of the trait genetic variation unexplained. Pathway- and network-based methodologies applied to GWAS aim to detect associations missed by standard single-marker approaches. The complex and non-random architecture of the genome makes it a challenge to derive an appropriate testing framework for such methodologies. We developed a rapid and simple permutation approach that uses GWAS SNP association results to establish the significance of pathway associations while accounting for the linkage disequilibrium structure of SNPs and the clustering of functionally related elements in the genome. All SNPs used in the GWAS are placed in a "circular genome" according to their location. Then the complete set of SNP association P values are permuted by rotation with respect to the genomic locations of the SNPs. Once these "simulated" P values are assigned, the joint gene P values are calculated using Fisher's combination test, and the association of pathways is tested using the hypergeometric test. The circular genomic permutation approach was applied to a human genome-wide association dataset. The data consists of 719 individuals from the ORCADES study genotyped for ~300,000 SNPs and measured for 51 traits ranging from physical to biochemical measurements. KEGG pathways (n = 225) were used as the sets of pathways to be tested. Our results demonstrate that the circular genomic permutations provide robust association P values. The non-permuted hypergeometric analysis generates ~1400 pathway-trait combination results with an association P value more significant than P ≤ 0.05, whereas applying circular genomic permutation reduces the number of significant results to a more credible 40% of that value. The circular permutation software ("genomicper") is available as an R package at http://cran.r-project.org/.
Keywords: GWAS; cardiac disease; genomicper R package; pathway-based; permutation method.
Figures
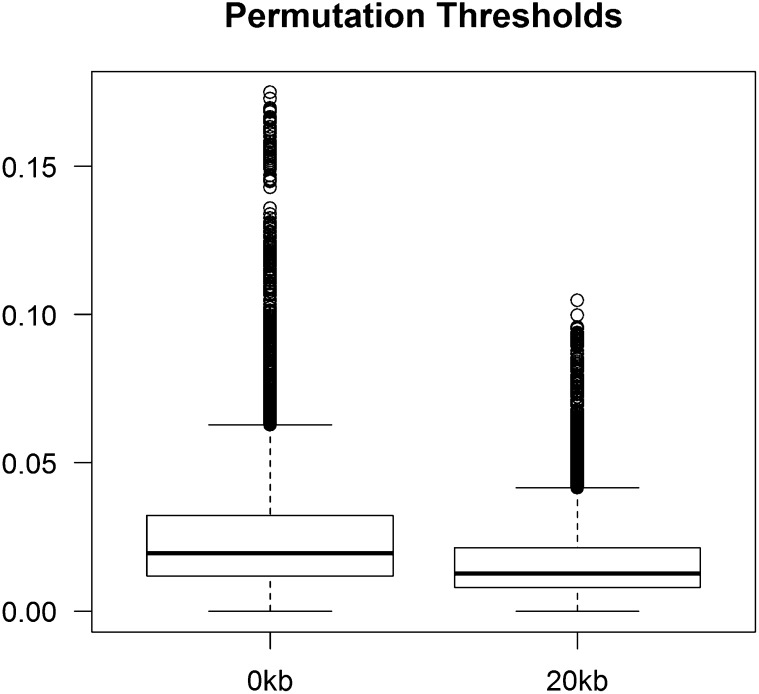
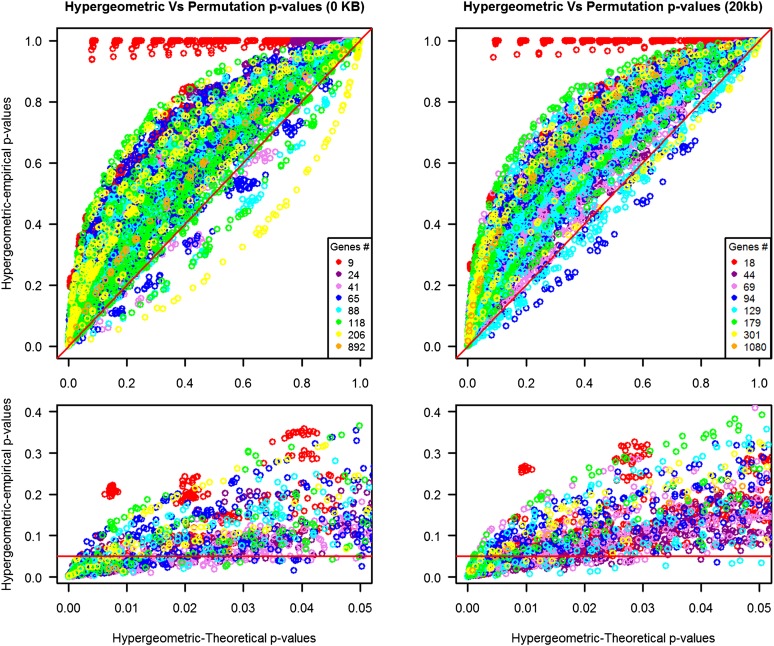
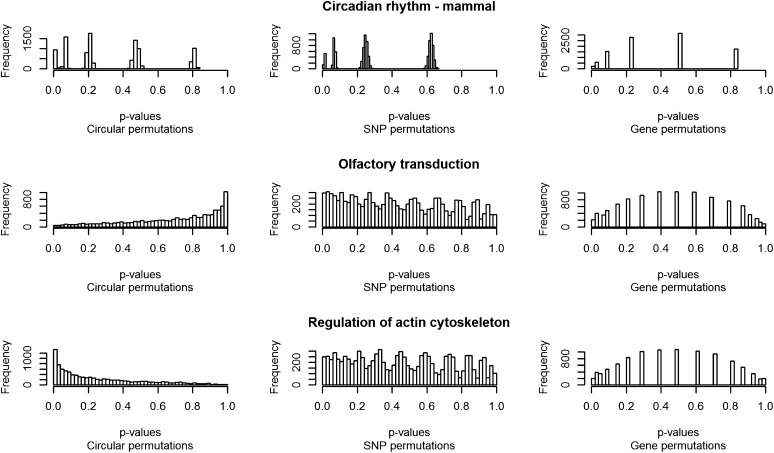
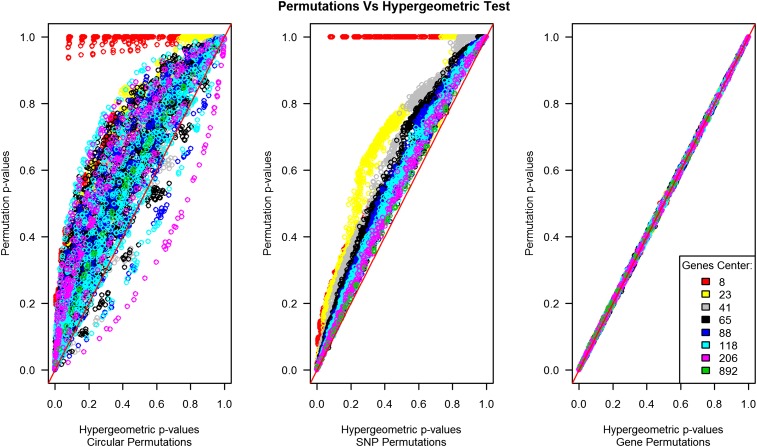
References
-
- Aulchenko Y. S., Ripke S., Isaacs A., van Duijn C. M., 2007. GenABEL: an R library for genome-wide association analysis. Bioinformatics 23: 1294–1296 - PubMed
-
- Callegaro A., Basso D., Bicciato S., 2006. A locally adaptive statistical procedure (LAP) to identify differentially expressed chromosomal regions. Bioinformatics 22: 2658–2666 - PubMed
-
- Caron H., Schaik B. v., Mee M. d., Baas F., Riggins G., et al. , 2001. The human transcriptome map: clustering of highly expressed genes in chromosomal domains. Science 291: 1289–1292 - PubMed
Publication types
MeSH terms
Grants and funding
- CZB/4/710/CSO_/Chief Scientist Office/United Kingdom
- MC_U127561128/MRC_/Medical Research Council/United Kingdom
- BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- MC_PC_U127592696/MRC_/Medical Research Council/United Kingdom
- MC_PC_U127561128/MRC_/Medical Research Council/United Kingdom
LinkOut - more resources
Full Text Sources