

Control of tumor bioenergetics and survival stress signaling by mitochondrial HSP90s
- PMID: 22975376
- PMCID: PMC3615709
- DOI: 10.1016/j.ccr.2012.07.015
Control of tumor bioenergetics and survival stress signaling by mitochondrial HSP90s
Abstract
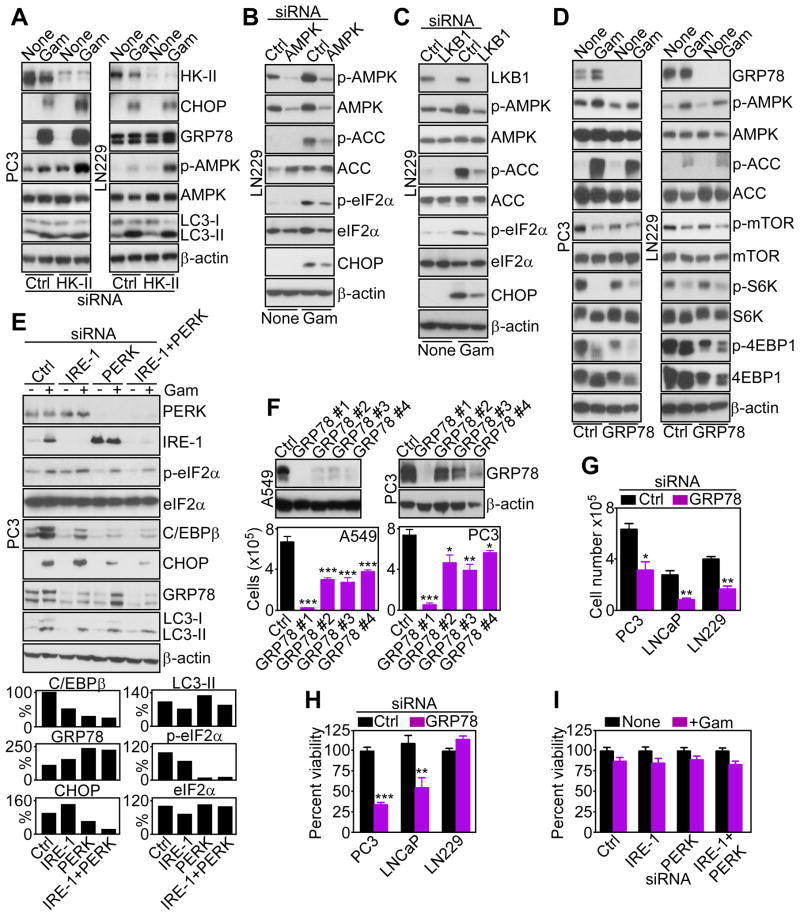
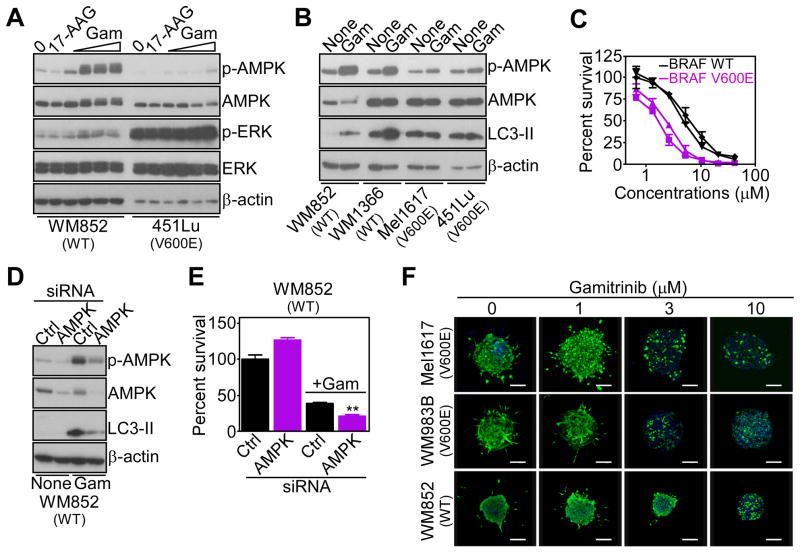
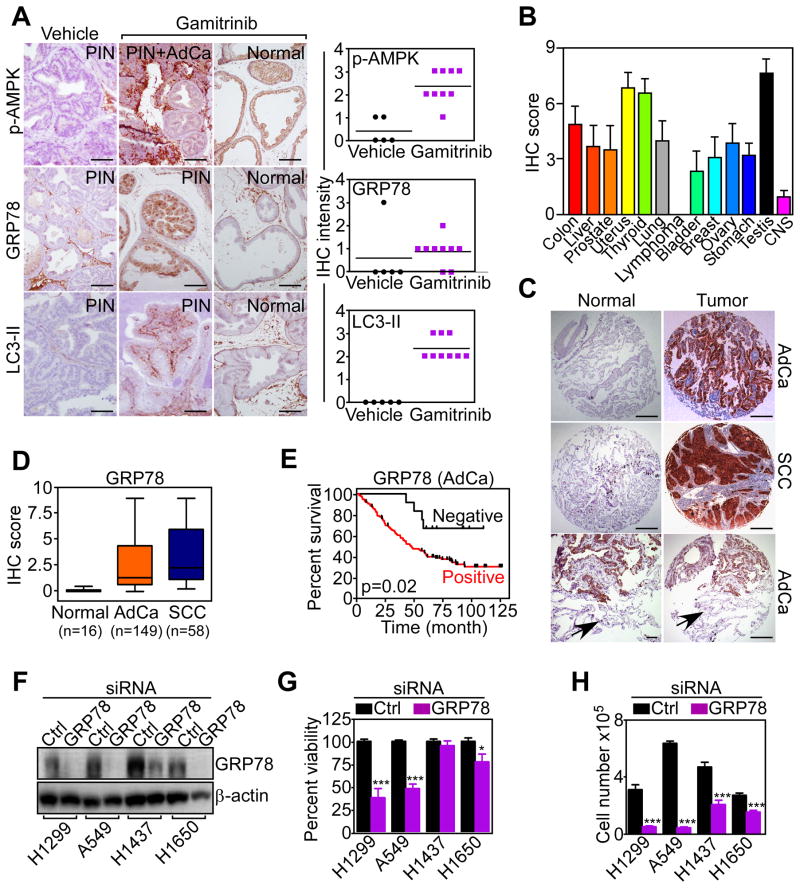
Tumors successfully adapt to constantly changing intra- and extracellular environments, but the wirings of this process are still largely elusive. Here, we show that heat-shock-protein-90-directed protein folding in mitochondria, but not cytosol, maintains energy production in tumor cells. Interference with this process activates a signaling network that involves phosphorylation of nutrient-sensing AMP-activated kinase, inhibition of rapamycin-sensitive mTOR complex 1, induction of autophagy, and expression of an endoplasmic reticulum unfolded protein response. This signaling network confers a survival and proliferative advantage to genetically disparate tumors, and correlates with worse outcome in lung cancer patients. Therefore, mitochondrial heat shock protein 90s are adaptive regulators of tumor bioenergetics and tractable targets for cancer therapy.
Copyright © 2012 Elsevier Inc. All rights reserved.
Conflict of interest statement
The authors declare no competing financial interest.
Figures
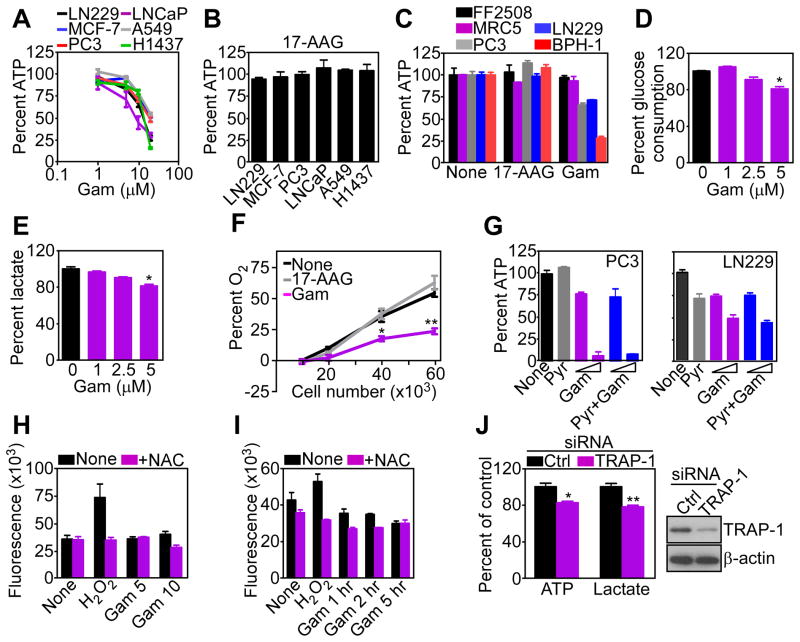
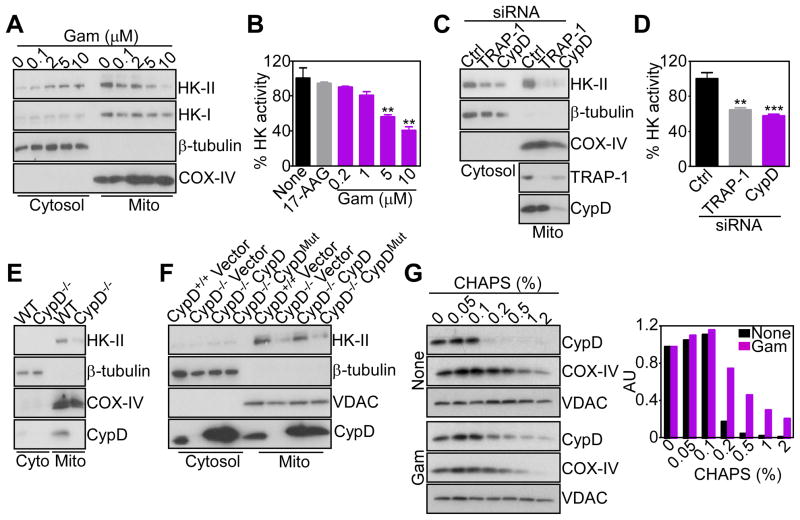
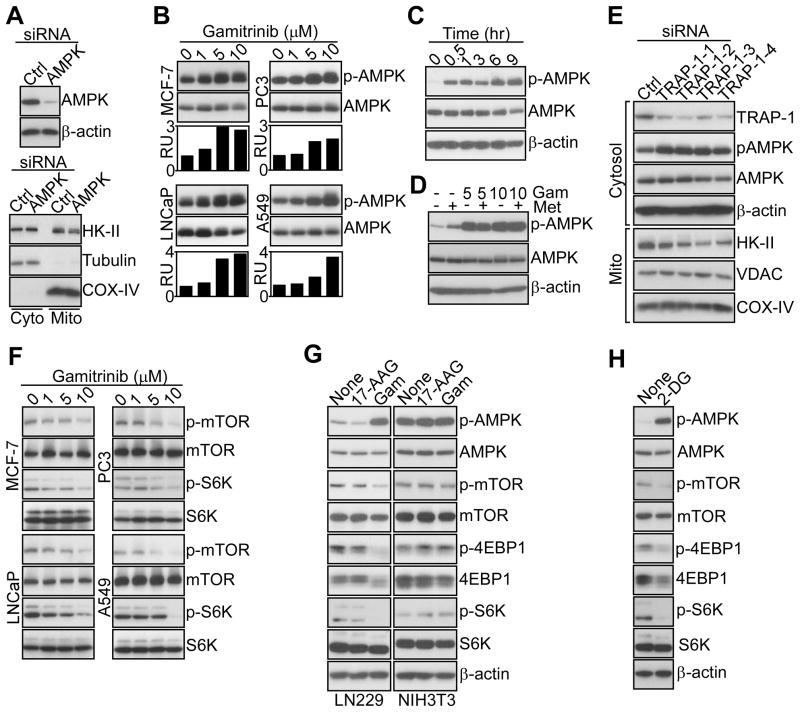
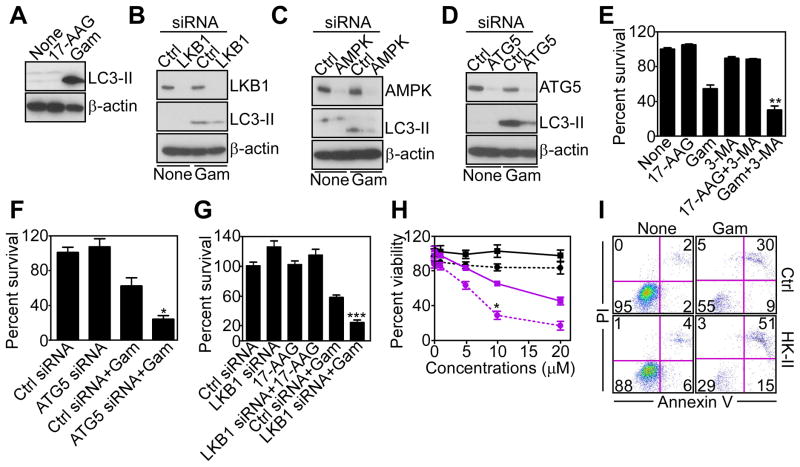

References
-
- Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, Brunskill EW, Sayen MR, Gottlieb RA, Dorn GW, et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature. 2005;434:658–662. - PubMed
-
- Green DR, Kroemer G. The pathophysiology of mitochondrial cell death. Science. 2004;305:626–629. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R01 CA078810/CA/NCI NIH HHS/United States
- R01 CA089720/CA/NCI NIH HHS/United States
- CA78810/CA/NCI NIH HHS/United States
- R37 HL054131/HL/NHLBI NIH HHS/United States
- P30 CA010815/CA/NCI NIH HHS/United States
- CA140043/CA/NCI NIH HHS/United States
- R01 CA109874/CA/NCI NIH HHS/United States
- CA010815/CA/NCI NIH HHS/United States
- R01 CA118005/CA/NCI NIH HHS/United States
- CA118005/CA/NCI NIH HHS/United States
- P01 CA140043/CA/NCI NIH HHS/United States
- HL54131/HL/NHLBI NIH HHS/United States
- R01 HL054131/HL/NHLBI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
