

Coevolution reveals a network of human proteins originating with multicellularity
- PMID: 22977115
- PMCID: PMC3548307
- DOI: 10.1093/molbev/mss218
Coevolution reveals a network of human proteins originating with multicellularity
Abstract
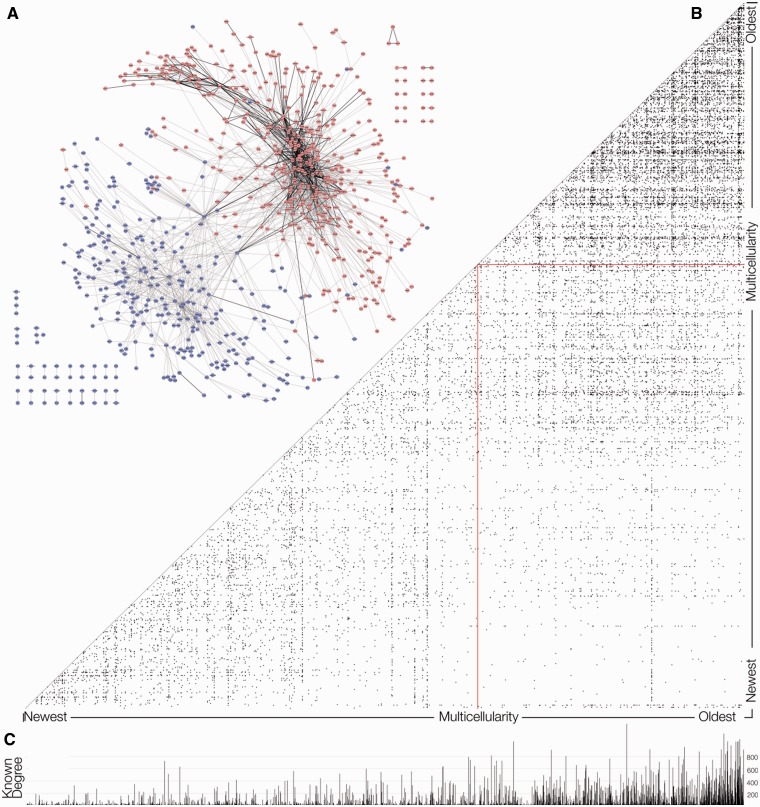
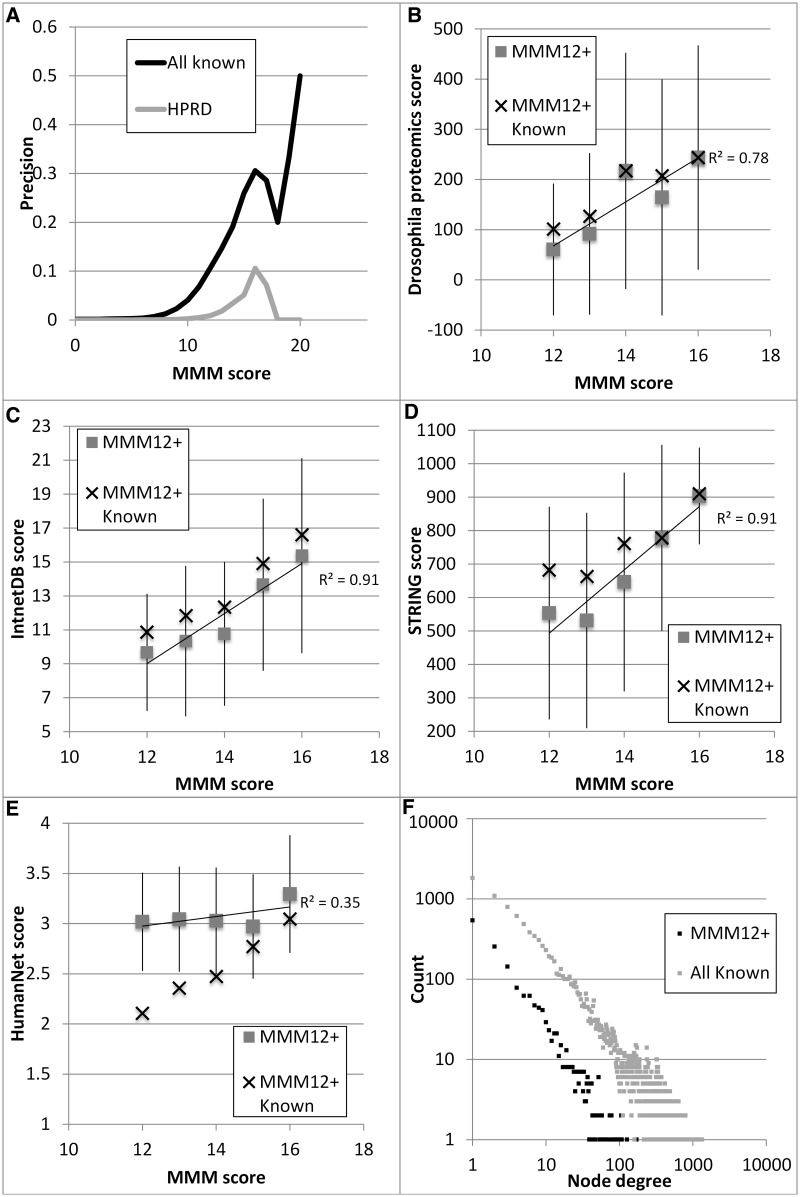
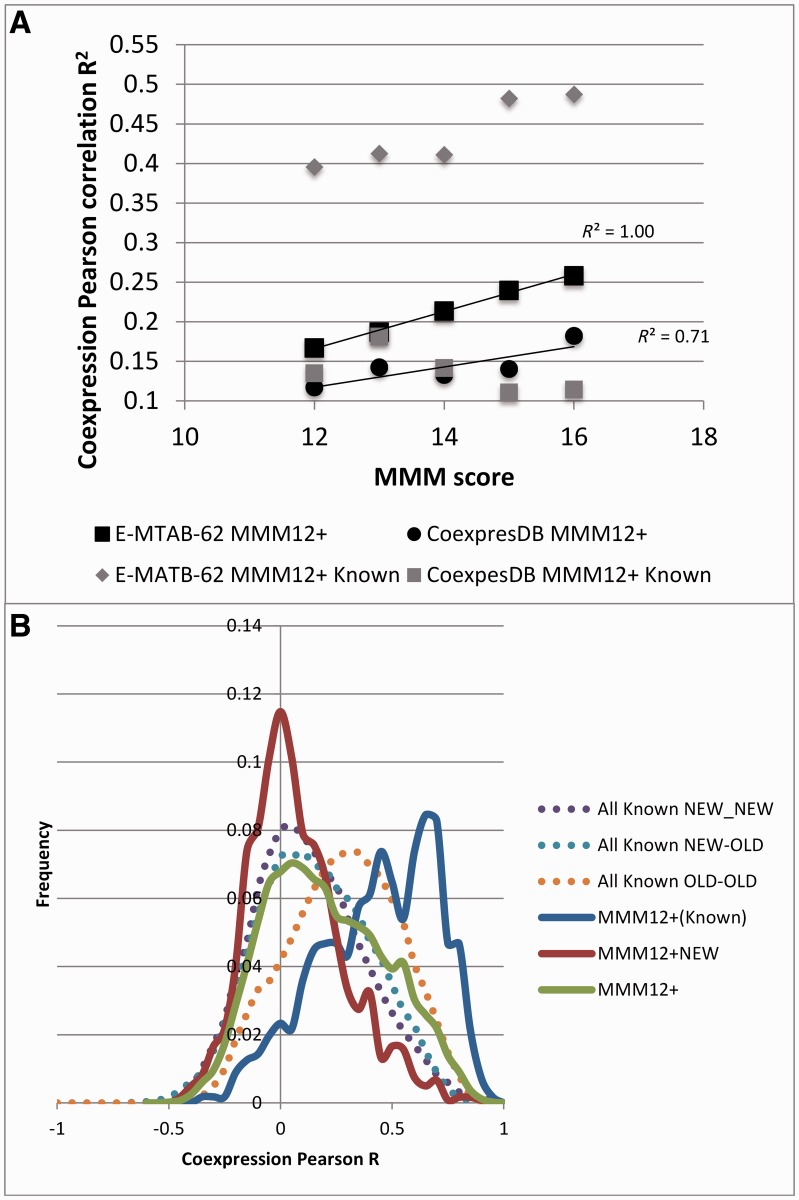
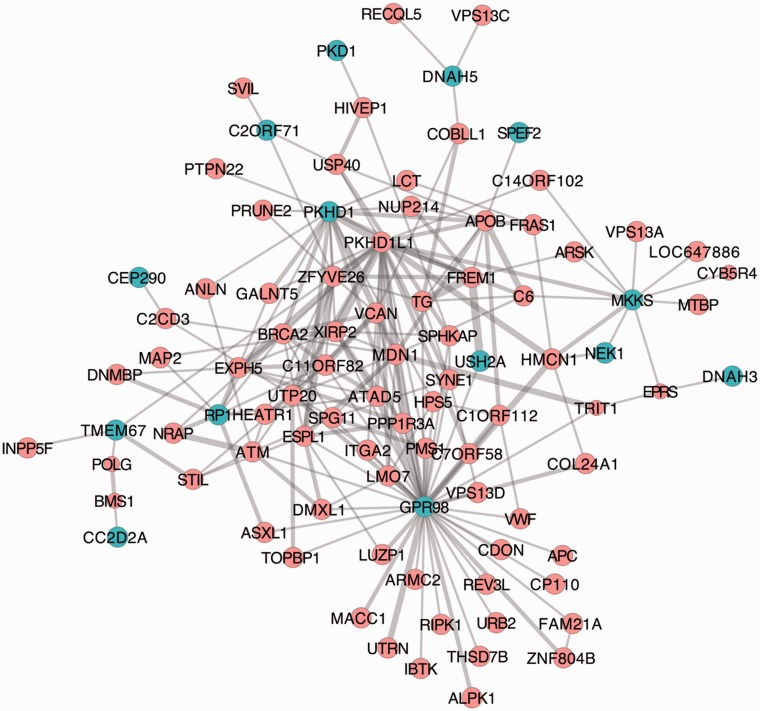
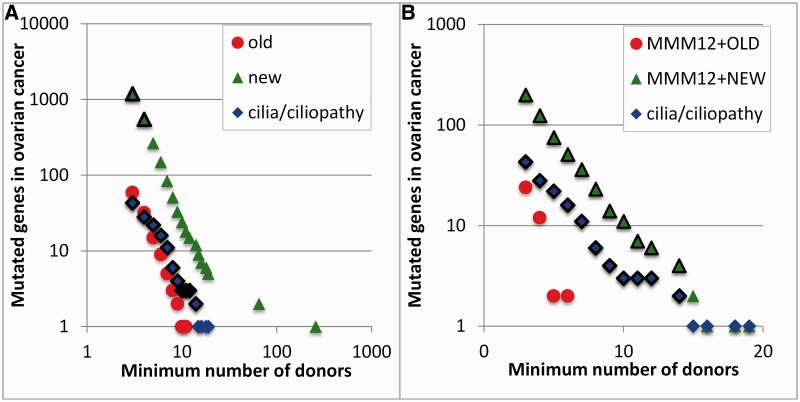
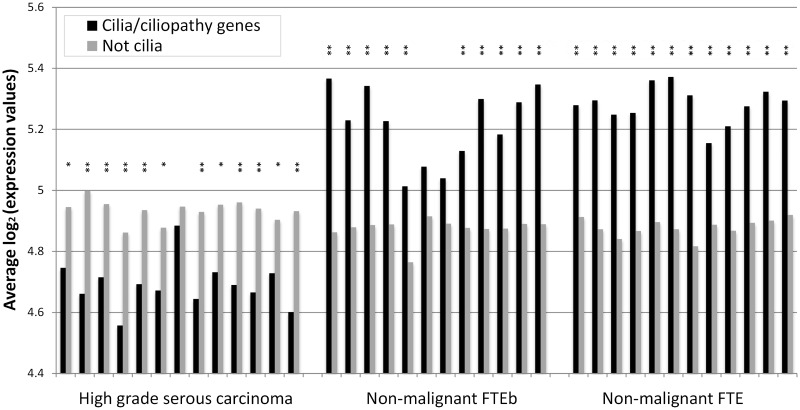
Protein interaction networks play central roles in biological systems, from simple metabolic pathways through complex programs permitting the development of organisms. Multicellularity could only have arisen from a careful orchestration of cellular and molecular roles and responsibilities, all properly controlled and regulated. Disease reflects a breakdown of this organismal homeostasis. To better understand the evolution of interactions whose dysfunction may be contributing factors to disease, we derived the human protein coevolution network using our MatrixMatchMaker algorithm and using the Orthologous MAtrix project (OMA) database as a source for protein orthologs from 103 eukaryotic genomes. We annotated the coevolution network using protein-protein interaction data, many functional data sources, and we explored the evolutionary rates and dates of emergence of the proteins in our data set. Strikingly, clustering based only on the topology of the coevolution network partitions it into two subnetworks, one generally representing ancient eukaryotic functions and the other functions more recently acquired during animal evolution. That latter subnetwork is enriched for proteins with roles in cell-cell communication, the control of cell division, and related multicellular functions. Further annotation using data from genetic disease databases and cancer genome sequences strongly implicates these proteins in both ciliopathies and cancer. The enrichment for such disease markers in the animal network suggests a functional link between these coevolving proteins. Genetic validation corroborates the recruitment of ancient cilia in the evolution of multicellularity.
Figures

References
-
- Alexa A, Rahnenführer J, Lengauer T. Improved scoring of functional groups from gene expression data by decorrelating GO graph structure. Bioinformatics. 2006;22:1600–1607. - PubMed
-
- Clark GW, Dar V, Bezginov A, Yang JM, Charlebois RL, Tillier ERM. Using coevolution to predict protein-protein interactions. Methods Mol Biol. 2011;781:237–256. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
