

Routine Access to Millisecond Time Scale Events with Accelerated Molecular Dynamics
- PMID: 22984356
- PMCID: PMC3438784
- DOI: 10.1021/ct300284c
Routine Access to Millisecond Time Scale Events with Accelerated Molecular Dynamics
Abstract
In this work, we critically assess the ability of the all-atom enhanced sampling method accelerated molecular dynamics (aMD) to investigate conformational changes in proteins that typically occur on the millisecond time scale. We combine aMD with the inherent power of graphics processor units (GPUs) and apply the implementation to the bovine pancreatic trypsin inhibitor (BPTI). A 500 ns aMD simulation is compared to a previous millisecond unbiased brute force MD simulation carried out on BPTI, showing that the same conformational space is sampled by both approaches. To our knowledge, this represents the first implementation of aMD on GPUs and also the longest aMD simulation of a biomolecule run to date. Our implementation is available to the community in the latest release of the Amber software suite (v12), providing routine access to millisecond events sampled from dynamics simulations using off the shelf hardware.
Figures
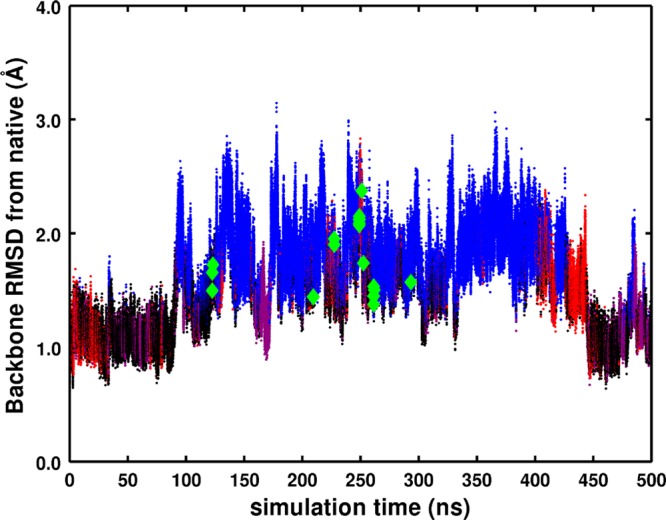
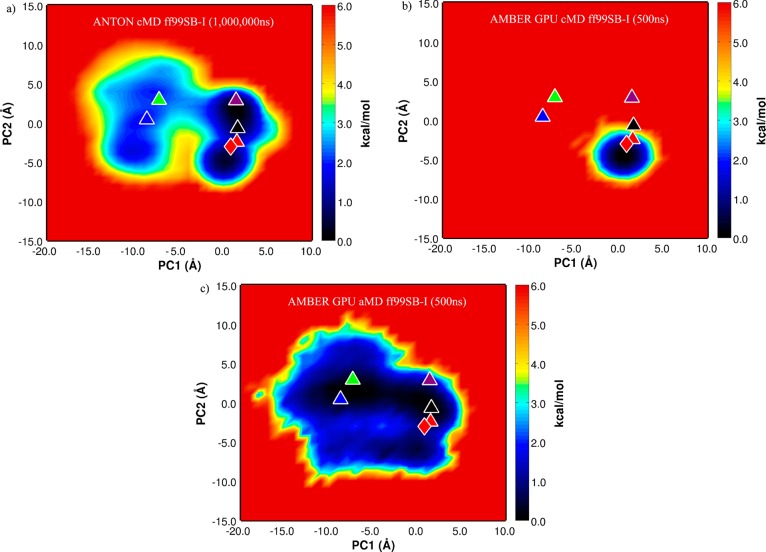
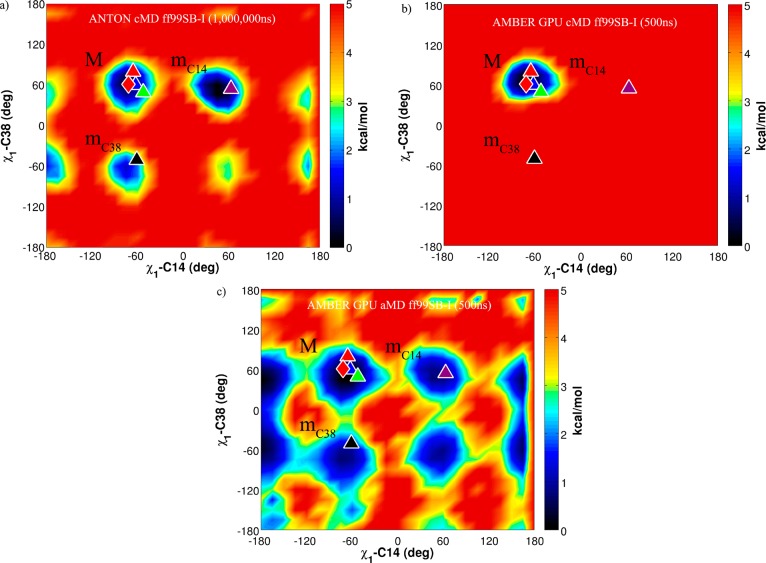
References
-
- Kubelka J.; Chiu T. K.; Davies D. R.; Eaton W. A.; Hofrichter J. Sub-microsecond protein folding. J. Mol. Biol. 2006, 359, 546–53. - PubMed
-
- Gilson M. K.; Zhou H. X. Calculation of protein-ligand binding affinities. Annu. Rev. Biophys. Biomol. Struct. 2007, 36, 21–42. - PubMed
-
- Lindahl E.; Sansom M. S. Membrane proteins: molecular dynamics simulations. Curr. Opin. Struct. Biol. 2008, 18, 425–31. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous