

A pipeline for automated annotation of yeast genome sequences by a conserved-synteny approach
- PMID: 22984983
- PMCID: PMC3507789
- DOI: 10.1186/1471-2105-13-237
A pipeline for automated annotation of yeast genome sequences by a conserved-synteny approach
Abstract
Background: Yeasts are a model system for exploring eukaryotic genome evolution. Next-generation sequencing technologies are poised to vastly increase the number of yeast genome sequences, both from resequencing projects (population studies) and from de novo sequencing projects (new species). However, the annotation of genomes presents a major bottleneck for de novo projects, because it still relies on a process that is largely manual.
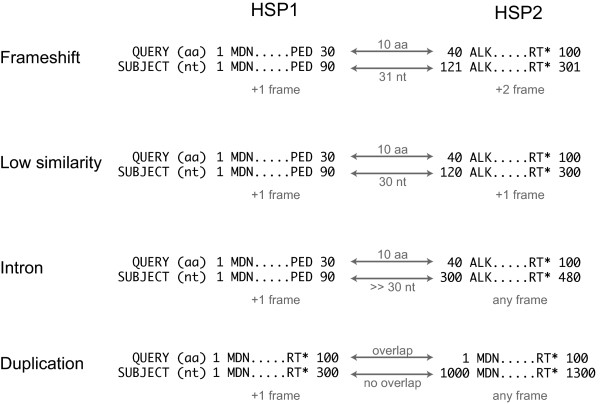
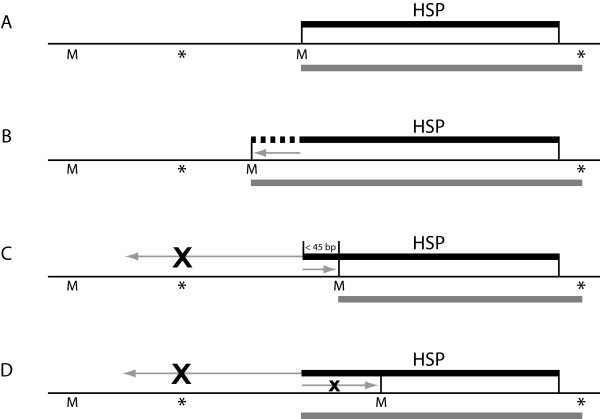
Results: Here we present the Yeast Genome Annotation Pipeline (YGAP), an automated system designed specifically for new yeast genome sequences lacking transcriptome data. YGAP does automatic de novo annotation, exploiting homology and synteny information from other yeast species stored in the Yeast Gene Order Browser (YGOB) database. The basic premises underlying YGAP's approach are that data from other species already tells us what genes we should expect to find in any particular genomic region and that we should also expect that orthologous genes are likely to have similar intron/exon structures. Additionally, it is able to detect probable frameshift sequencing errors and can propose corrections for them. YGAP searches intelligently for introns, and detects tRNA genes and Ty-like elements.
Conclusions: In tests on Saccharomyces cerevisiae and on the genomes of Naumovozyma castellii and Tetrapisispora blattae newly sequenced with Roche-454 technology, YGAP outperformed another popular annotation program (AUGUSTUS). For S. cerevisiae and N. castellii, 91-93% of YGAP's predicted gene structures were identical to those in previous manually curated gene sets. YGAP has been implemented as a webserver with a user-friendly interface at http://wolfe.gen.tcd.ie/annotation.
Figures

Similar articles
-
Visualizing syntenic relationships among the hemiascomycetes with the Yeast Gene Order Browser.Nucleic Acids Res. 2006 Jan 1;34(Database issue):D452-5. doi: 10.1093/nar/gkj041. Nucleic Acids Res. 2006. PMID: 16381909 Free PMC article.
-
The Yeast Gene Order Browser: combining curated homology and syntenic context reveals gene fate in polyploid species.Genome Res. 2005 Oct;15(10):1456-61. doi: 10.1101/gr.3672305. Epub 2005 Sep 16. Genome Res. 2005. PMID: 16169922 Free PMC article.
-
Filling annotation gaps in yeast genomes using genome-wide contact maps.Bioinformatics. 2014 Aug 1;30(15):2105-13. doi: 10.1093/bioinformatics/btu162. Epub 2014 Apr 7. Bioinformatics. 2014. PMID: 24711652
-
Comparative study on synteny between yeasts and vertebrates.C R Biol. 2011 Aug-Sep;334(8-9):629-38. doi: 10.1016/j.crvi.2011.05.011. Epub 2011 Jul 5. C R Biol. 2011. PMID: 21819944 Review.
-
Yeasty clocks: dating genomic changes in yeasts.C R Biol. 2011 Aug-Sep;334(8-9):620-8. doi: 10.1016/j.crvi.2011.05.010. Epub 2011 Jul 1. C R Biol. 2011. PMID: 21819943 Review.
Cited by
-
Comparative Genomic and Transcriptomic Analysis Reveals Specific Features of Gene Regulation in Kluyveromyces marxianus.Front Microbiol. 2021 Feb 26;12:598060. doi: 10.3389/fmicb.2021.598060. eCollection 2021. Front Microbiol. 2021. PMID: 33717000 Free PMC article.
-
Landscape of the Dark Transcriptome Revealed Through Re-mining Massive RNA-Seq Data.Front Genet. 2021 Aug 16;12:722981. doi: 10.3389/fgene.2021.722981. eCollection 2021. Front Genet. 2021. PMID: 34484307 Free PMC article.
-
Ploidy evolution in a wild yeast is linked to an interaction between cell type and metabolism.PLoS Biol. 2023 Nov 9;21(11):e3001909. doi: 10.1371/journal.pbio.3001909. eCollection 2023 Nov. PLoS Biol. 2023. PMID: 37943740 Free PMC article.
-
Draft Genome Sequences of Three Clinical Isolates of the Pathogenic Yeast Candida glabrata.Microbiol Resour Announc. 2019 Aug 29;8(35):e00278-19. doi: 10.1128/MRA.00278-19. Microbiol Resour Announc. 2019. PMID: 31467089 Free PMC article.
-
Genome sequence data of the antagonistic soil-borne yeast Cyberlindnera sargentensis (SHA 17.2).Data Brief. 2022 Jan 5;40:107799. doi: 10.1016/j.dib.2022.107799. eCollection 2022 Feb. Data Brief. 2022. PMID: 35071701 Free PMC article.
References
-
- Dujon B. Yeast evolutionary genomics. Nat Rev Genet. 2010;11:512–524. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
