

Missense mutations in ITPR1 cause autosomal dominant congenital nonprogressive spinocerebellar ataxia
- PMID: 22986007
- PMCID: PMC3545966
- DOI: 10.1186/1750-1172-7-67
Missense mutations in ITPR1 cause autosomal dominant congenital nonprogressive spinocerebellar ataxia
Erratum in
-
Correction to: Missense mutations in ITPR1 cause autosomal dominant congenital nonprogressive spinocerebellar ataxia.Orphanet J Rare Dis. 2022 Mar 29;17(1):143. doi: 10.1186/s13023-022-02297-7. Orphanet J Rare Dis. 2022. PMID: 35351177 Free PMC article. No abstract available.
Abstract
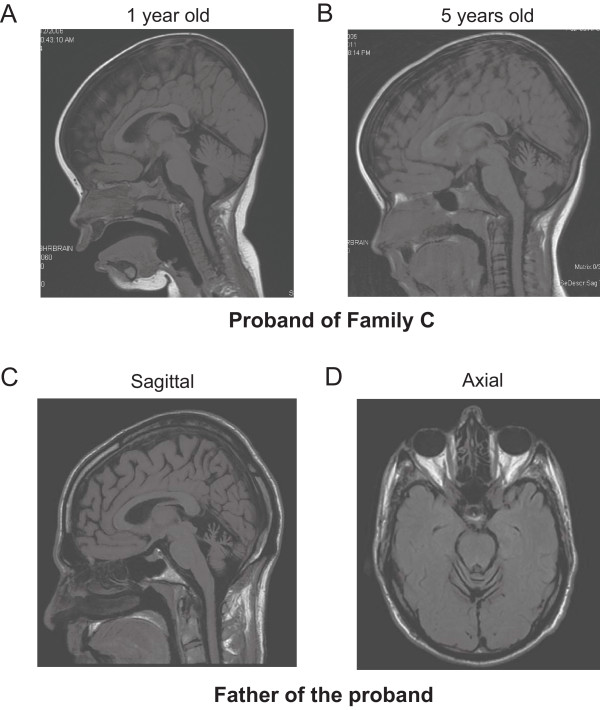
Background: Congenital nonprogressive spinocerebellar ataxia is characterized by early gross motor delay, hypotonia, gait ataxia, mild dysarthria and dysmetria. The clinical presentation remains fairly stable and may be associated with cerebellar atrophy. To date, only a few families with autosomal dominant congenital nonprogressive spinocerebellar ataxia have been reported. Linkage to 3pter was demonstrated in one large Australian family and this locus was designated spinocerebellar ataxia type 29. The objective of this study is to describe an unreported Canadian family with autosomal dominant congenital nonprogressive spinocerebellar ataxia and to identify the underlying genetic causes in this family and the original Australian family.
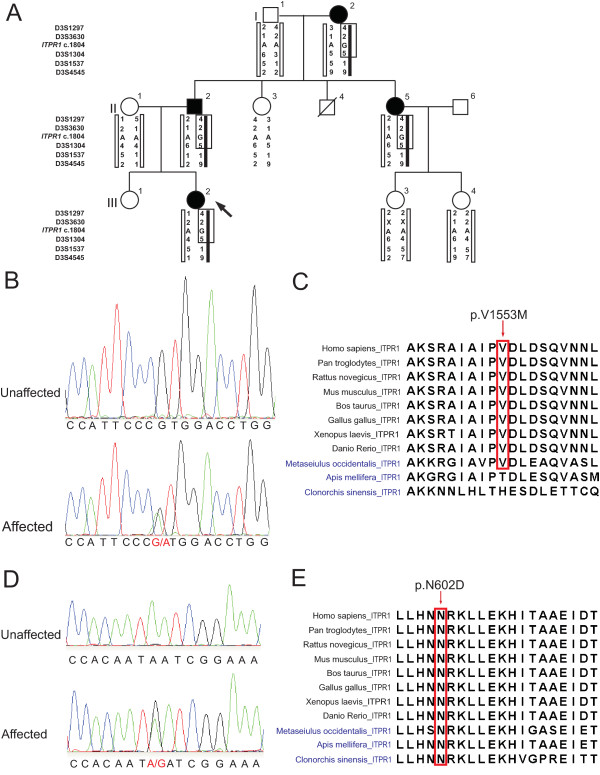
Methods and results: Exome sequencing was performed for the Australian family, resulting in the identification of a heterozygous mutation in the ITPR1 gene. For the Canadian family, genotyping with microsatellite markers and Sanger sequencing of ITPR1 gene were performed; a heterozygous missense mutation in ITPR1 was identified.
Conclusions: ITPR1 encodes inositol 1,4,5-trisphosphate receptor, type 1, a ligand-gated ion channel that mediates calcium release from the endoplasmic reticulum. Deletions of ITPR1 are known to cause spinocerebellar ataxia type 15, a distinct and very slowly progressive form of cerebellar ataxia with onset in adulthood. Our study demonstrates for the first time that, in addition to spinocerebellar ataxia type 15, alteration of ITPR1 function can cause a distinct congenital nonprogressive ataxia; highlighting important clinical heterogeneity associated with the ITPR1 gene and a significant role of the ITPR1-related pathway in the development and maintenance of the normal functions of the cerebellum.
Figures
References
-
- Harding AE. Classification of the hereditary ataxias and paraplegias. Lancet. 1983;1:1151–1155. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
