

Towards a new tuberculosis drug: pyridomycin - nature's isoniazid
- PMID: 22987724
- PMCID: PMC3491834
- DOI: 10.1002/emmm.201201689
Towards a new tuberculosis drug: pyridomycin - nature's isoniazid
Abstract
Tuberculosis, a global threat to public health, is becoming untreatable due to widespread drug resistance to frontline drugs such as the InhA-inhibitor isoniazid. Historically, by inhibiting highly vulnerable targets, natural products have been an important source of antibiotics including potent anti-tuberculosis agents. Here, we describe pyridomycin, a compound produced by Dactylosporangium fulvum with specific cidal activity against mycobacteria. By selecting pyridomycin-resistant mutants of Mycobacterium tuberculosis, whole-genome sequencing and genetic validation, we identified the NADH-dependent enoyl- (Acyl-Carrier-Protein) reductase InhA as the principal target and demonstrate that pyridomycin inhibits mycolic acid synthesis in M. tuberculosis. Furthermore, biochemical and structural studies show that pyridomycin inhibits InhA directly as a competitive inhibitor of the NADH-binding site, thereby identifying a new, druggable pocket in InhA. Importantly, the most frequently encountered isoniazid-resistant clinical isolates remain fully susceptible to pyridomycin, thus opening new avenues for drug development. →See accompanying article http://dx.doi.org/10.1002/emmm.201201811.
Copyrights © 2012 The Authors. Published by John Wiley and Sons, Ltd on behalf of EMBO.
Figures
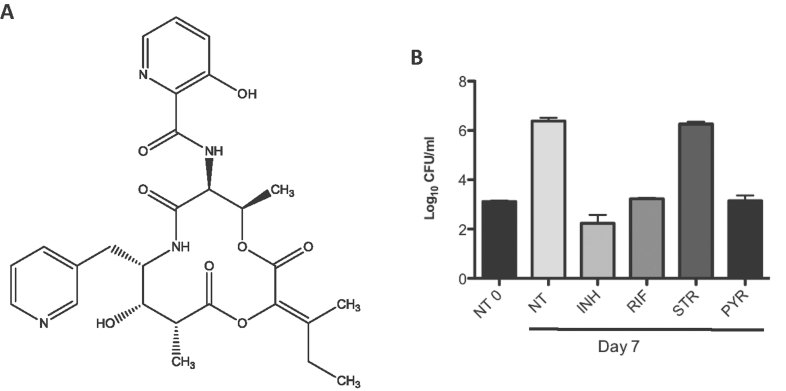
Chemical structure of pyridomycin.
The activity of pyridomycin on intracellular M. tuberculosis was tested in activated THP-1-derived macrophages. Cells were infected at an MOI of 1:1 with M. tuberculosis Erdman and treated with isoniazid (INH) at 1 µg/ml, rifampicin (RIF) at 1 µg/ml, streptomycin (STR) at 10 µg/ml or pyridomycin (PYR) at 10 µg/ml. Colony forming units (CFU) were determined after 7 days exposure to drugs. NT refers to the untreated sample and NT0 to untreated sample at time 0. The experiment was performed in duplicate and results are shown as mean values and standard errors.
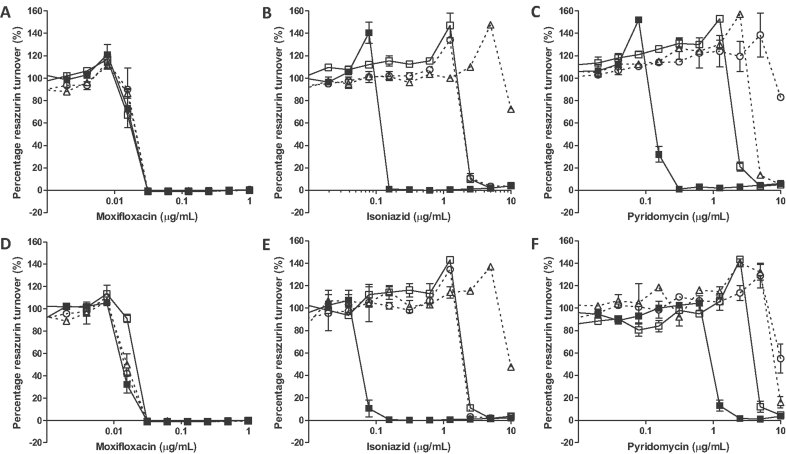
A-C. The compound susceptibility of wild-type H37Rv transformed with the control vector pMV261 (filled squares), pMVinhA (open squares), pMVinhA (S94A) (open triangle) or pMVinhA (D148G) (open circle) to: (A) moxifloxacin, (B) isoniazid or (C) pyridomycin.
D-F. The compound susceptibility of pyridomycin-resistant mutant PYR7 transformed with the control vector pMV261 (filled squares), pMVinhA (open squares), pMVinhA (S94A) (open triangle) or pMVinhA (D148G) (open circle) to: (D) moxifloxacin, (E) isoniazid or (F) pyridomycin.
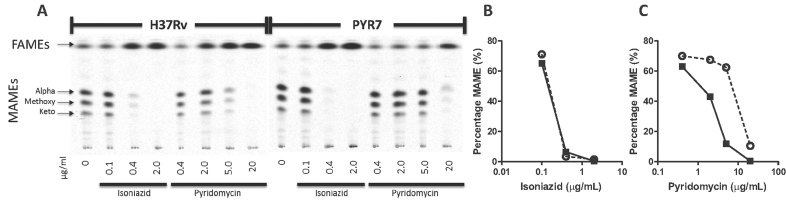
A. 14C-labeled FAMEs and MAMEs were separated by thin-layer chromatography and detected by autoradiography.
B,C. Quantification of the MAME band intensity relative to the density of the FAMEs illustrates the inhibition of MAME synthesis by pyridomycin (B) and isoniazid (C) in H37Rv (black squares) and PYR7 (open circles).
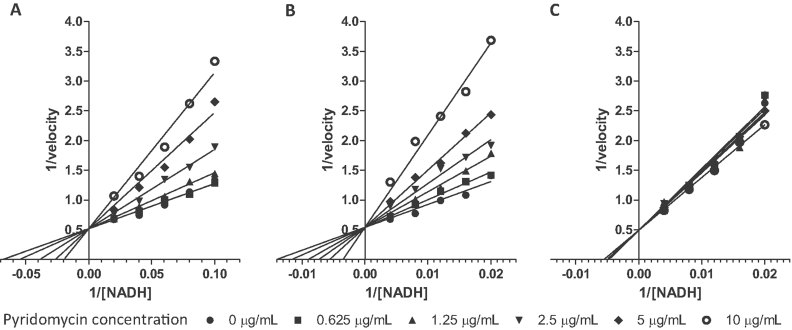
A-C. Lineweaver–Burk plot showing the competitive inhibition of wild-type InhA (A), InhA (S94A) (B) and InhA (D148G) (C) by pyridomycin in the presence of NADH.
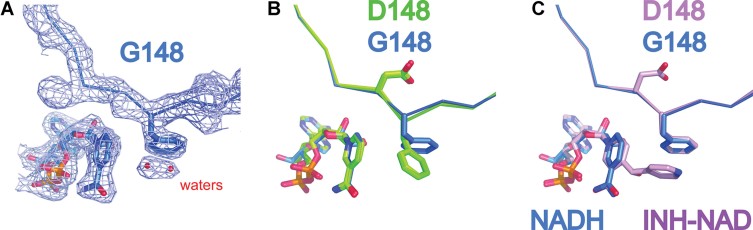
The 2fo-fc electron density map at 1 sigma of InhA (D148G):NADH structure (at 2.45 Å) with Phe149 and NADH represented as sticks.
Superposition of InhA (D148G):NADH (blue) on InhA:NADH and InhA (S94A):NADH (both in green) clearly shows the D148G mutation and the resulting 90° rotation of Phe149.
Overlay of InhA (D148G):NADH (Blue) and InhA:NAD-INH (PDB code 1Z1D: Pink) shows that, in this case, Phe149 occupies the same position and explains why InhA (D148G) is still sensitive to inhibition by isoniazid.
References
-
- Baldock C, Rafferty JB, Sedelnikova SE, Baker PJ, Stuitje AR, Slabas AR, Hawkes TR, Rice DW. A mechanism of drug action revealed by structural studies of enoyl reductase. Science. 1996;274:2107–2110. - PubMed
-
- Banerjee A, Dubnau E, Quemard A, Balasubramanian V, Um KS, Wilson T, Collins D, de Lisle G, Jacobs WR., Jr inhA, a gene encoding a target for isoniazid and ethionamide in Mycobacterium tuberculosis. Science. 1994;263:227–230. - PubMed
-
- Cegielski JP. Extensively drug-resistant tuberculosis: “There must be some kind of way out of here”. Clin Infect Dis. 2010;50:S195–S200. - PubMed
-
- Cole ST, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, Gordon SV, Eiglmeier K, Gas S, Barry CE, III, et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 1998;393:537–544. - PubMed
-
- Cole ST, Riccardi G. New tuberculosis drugs on the horizon. Curr Opin Microbiol. 2011;14:570–576. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
