

Isozyme-specific interaction of protein kinase Cδ with mitochondria dissected using live cell fluorescence imaging
- PMID: 22988234
- PMCID: PMC3488061
- DOI: 10.1074/jbc.M112.412635
Isozyme-specific interaction of protein kinase Cδ with mitochondria dissected using live cell fluorescence imaging
Abstract
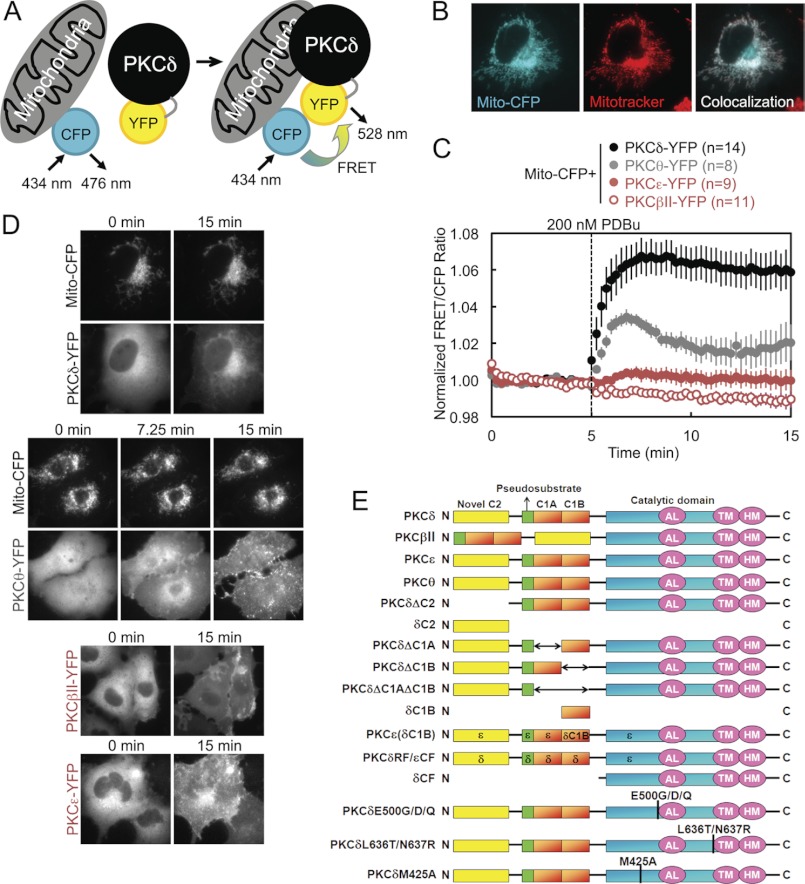
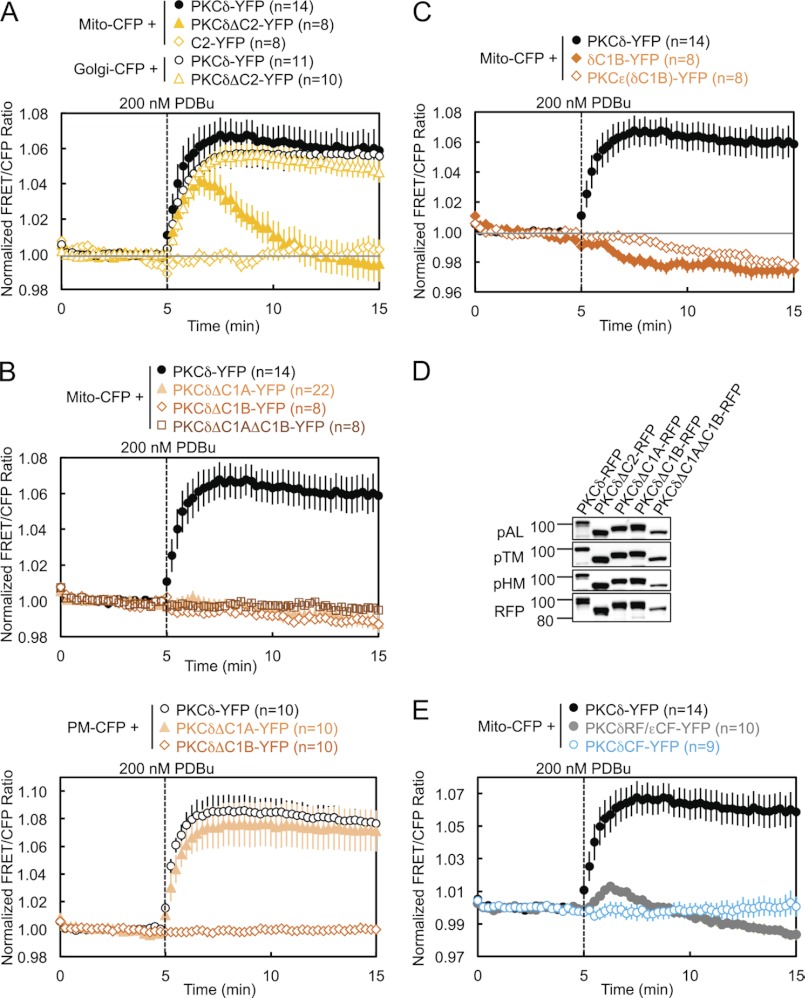
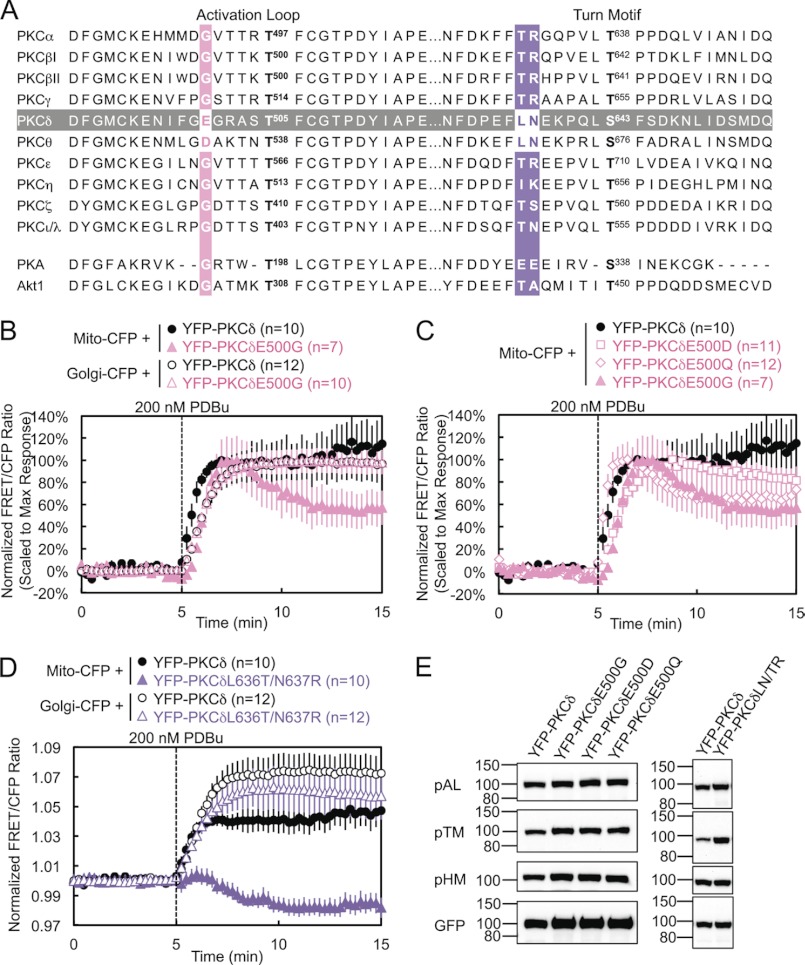
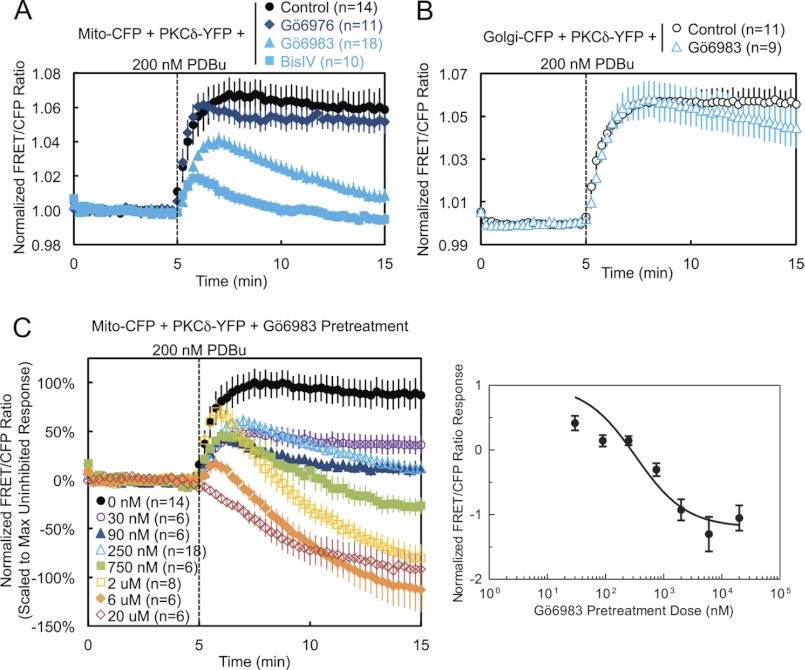
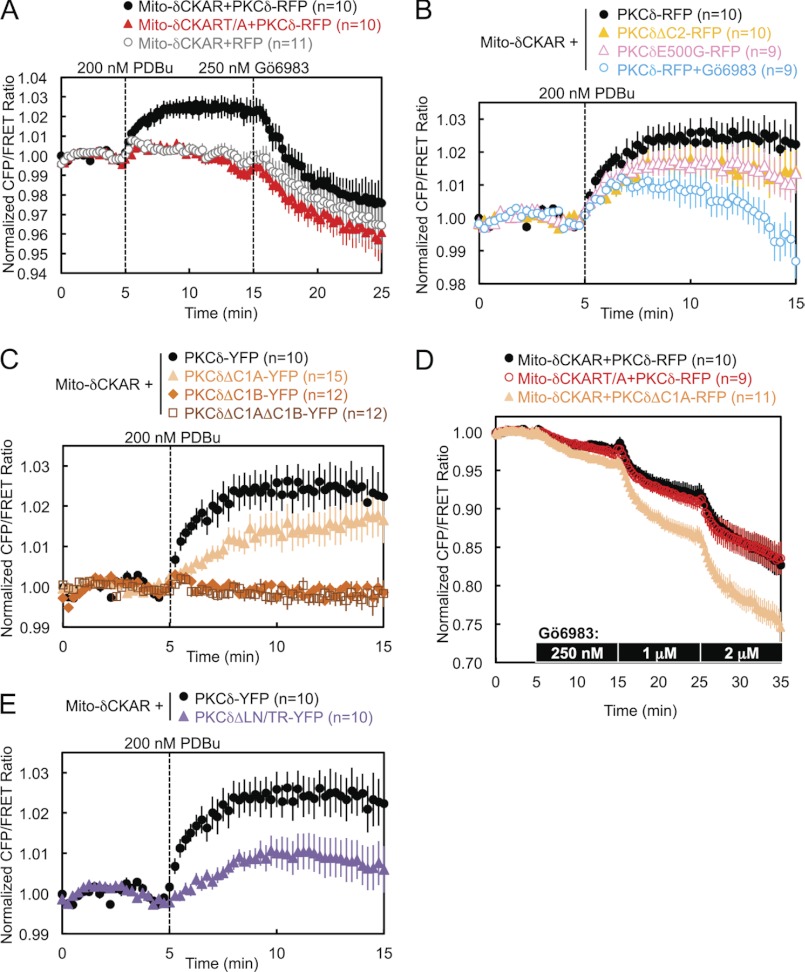
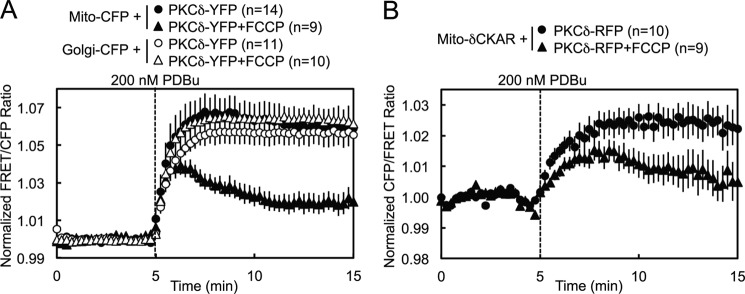
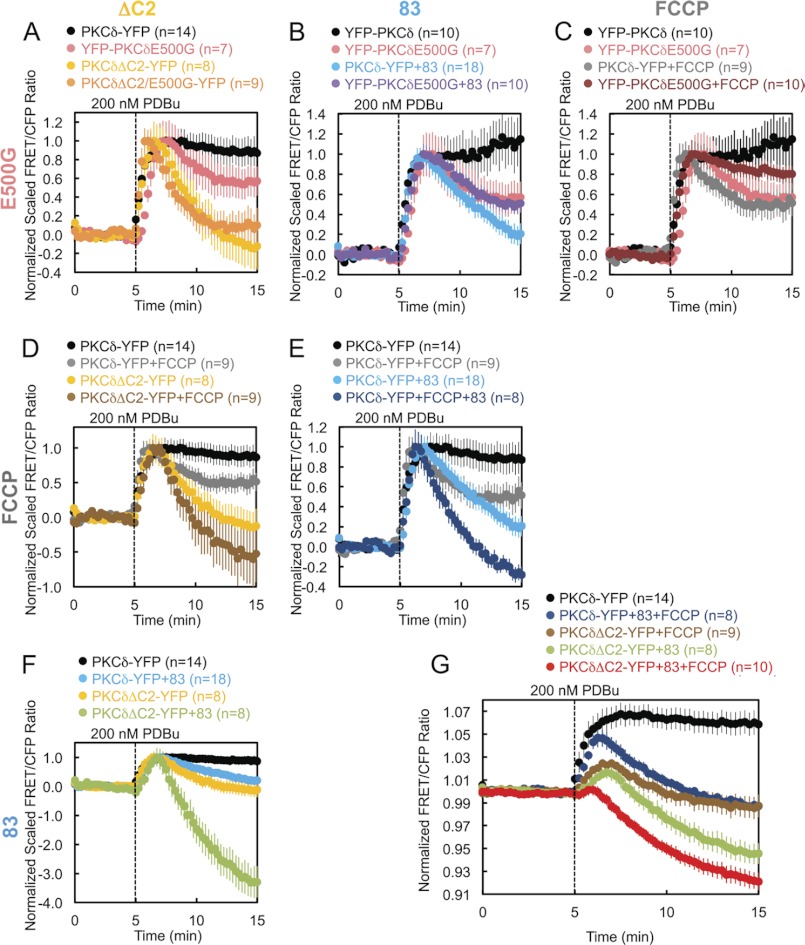
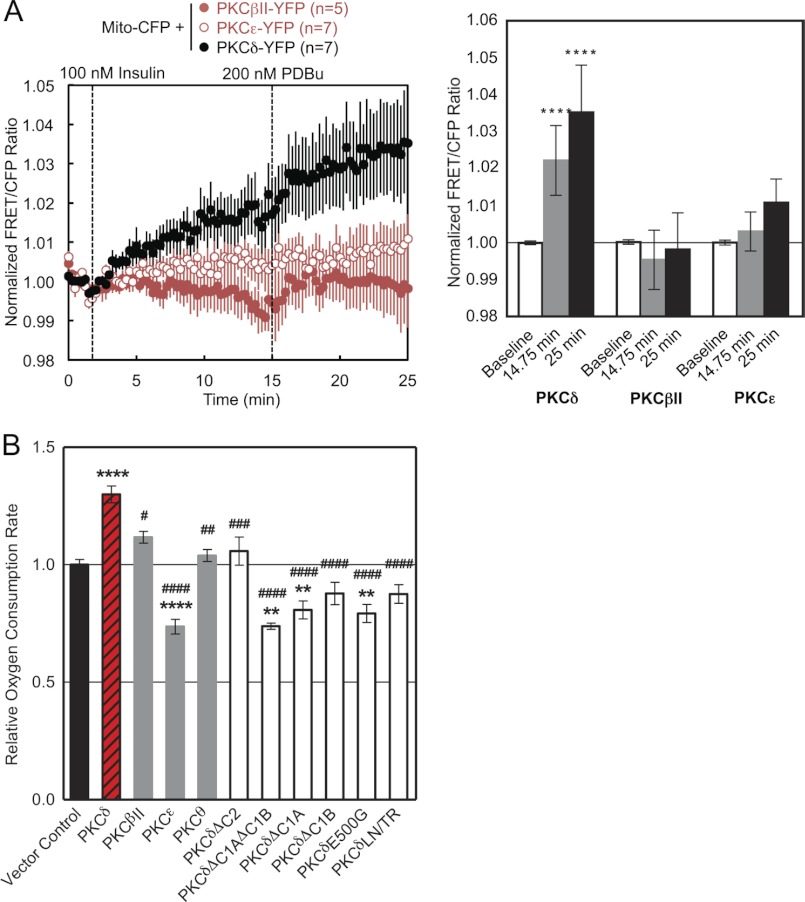
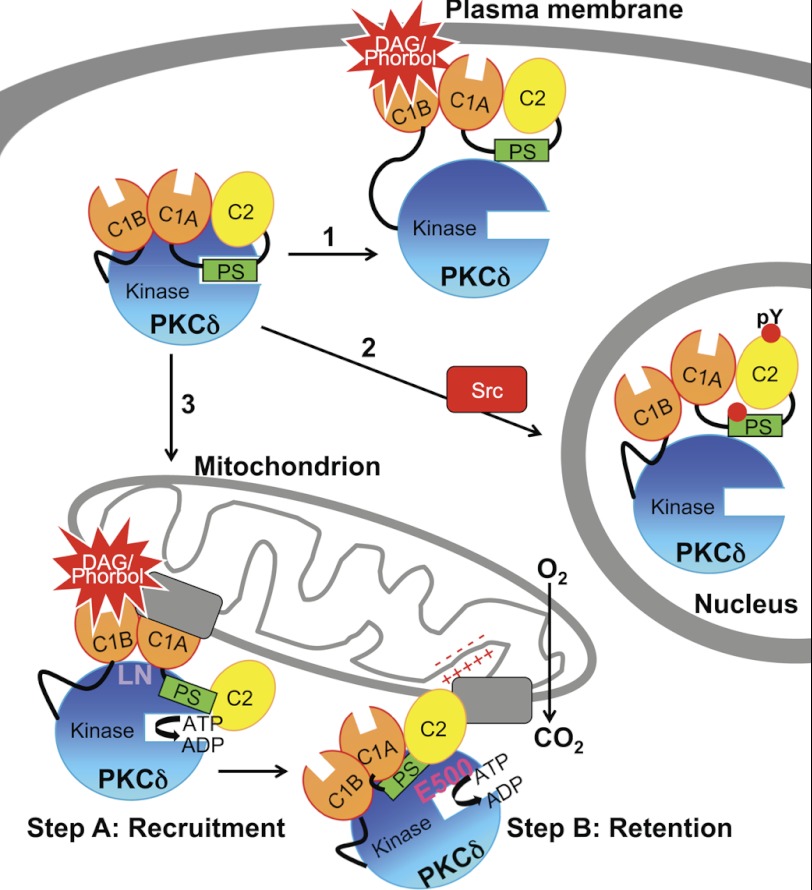
PKCδ signaling to mitochondria has been implicated in both mitochondrial apoptosis and metabolism. However, the mechanism by which PKCδ interacts with mitochondria is not well understood. Using FRET-based imaging, we show that PKCδ interacts with mitochondria by a novel and isozyme-specific mechanism distinct from its canonical recruitment to other membranes such as the plasma membrane or Golgi. Specifically, we show that PKCδ interacts with mitochondria following stimulation with phorbol esters or, in L6 myocytes, with insulin via a mechanism that requires two steps. In the first step, PKCδ translocates acutely to mitochondria by a mechanism that requires its C1A and C1B domains and a Leu-Asn sequence in its turn motif. In the second step, PKCδ is retained at mitochondria by a mechanism that depends on its C2 domain, a unique Glu residue in its activation loop, intrinsic catalytic activity, and the mitochondrial membrane potential. In contrast, of these determinants, only the C1B domain is required for the phorbol ester-stimulated translocation of PKCδ to other membranes. PKCδ also basally localizes to mitochondria and increases mitochondrial respiration via many of the same determinants that promote its agonist-evoked interaction. PKCδ localized to mitochondria has robust activity, as revealed by a FRET reporter of PKCδ-specific activity (δCKAR). These data support a model in which multiple determinants unique to PKCδ drive a specific interaction with mitochondria that promotes mitochondrial respiration.
Figures

References
-
- Griner E. M., Kazanietz M. G. (2007) Protein kinase C and other diacylglycerol effectors in cancer. Nat. Rev. Cancer 7, 281–294 - PubMed
-
- Rosse C., Linch M., Kermorgant S., Cameron A. J., Boeckeler K., Parker P. J. (2010) PKC and the control of localized signal dynamics. Nat. Rev. Mol. Cell Biol. 11, 103–112 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
