

The EGF receptor and HER2 participate in TNF-α-dependent MAPK activation and IL-8 secretion in intestinal epithelial cells
- PMID: 22988345
- PMCID: PMC3440955
- DOI: 10.1155/2012/207398
The EGF receptor and HER2 participate in TNF-α-dependent MAPK activation and IL-8 secretion in intestinal epithelial cells
Abstract
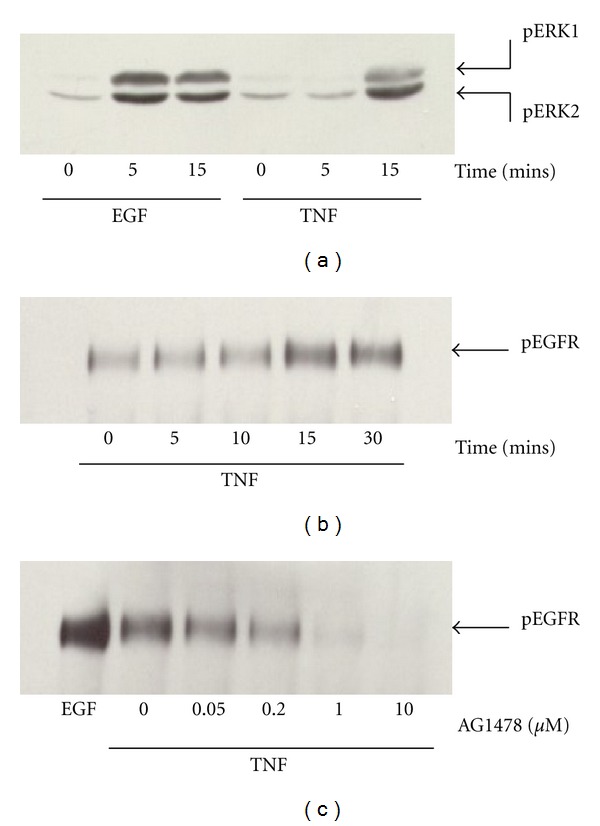
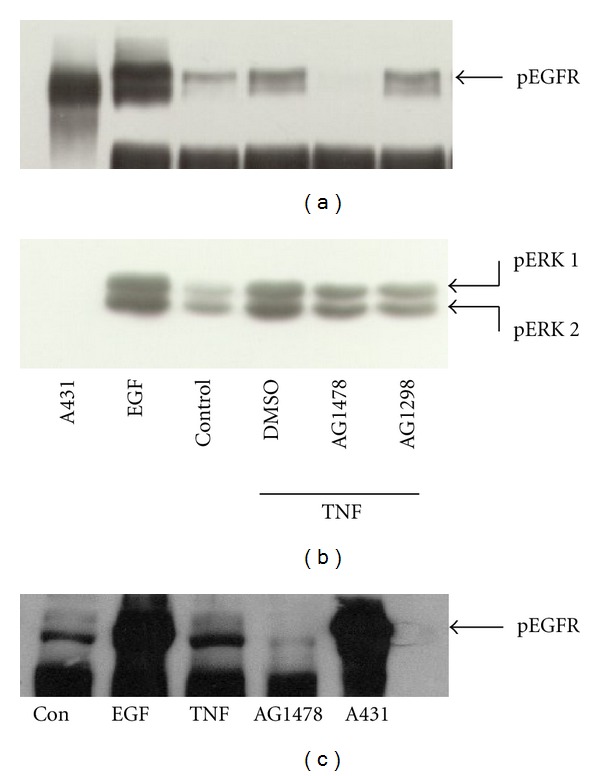
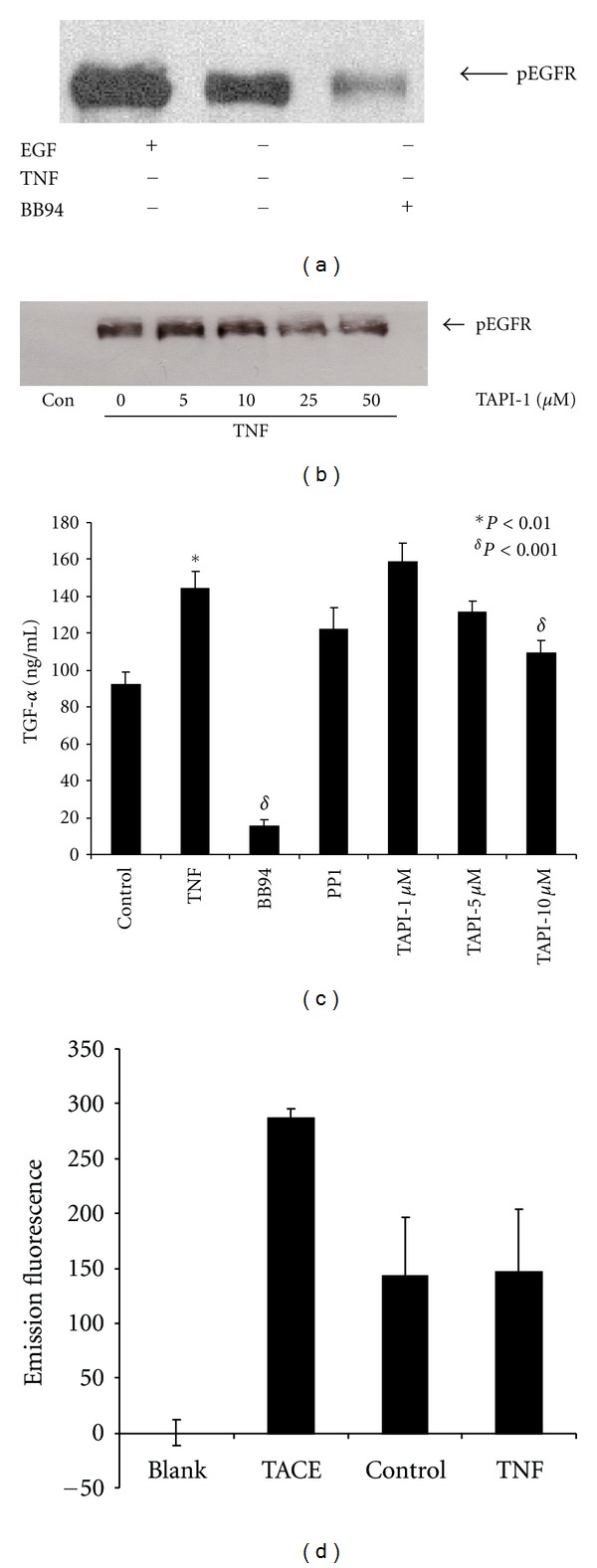
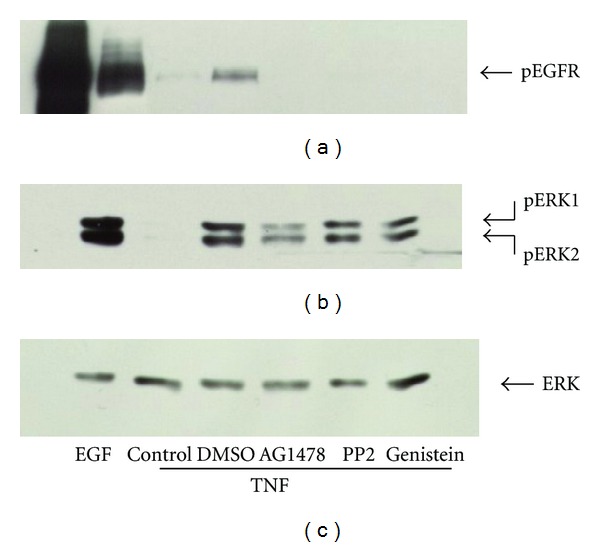
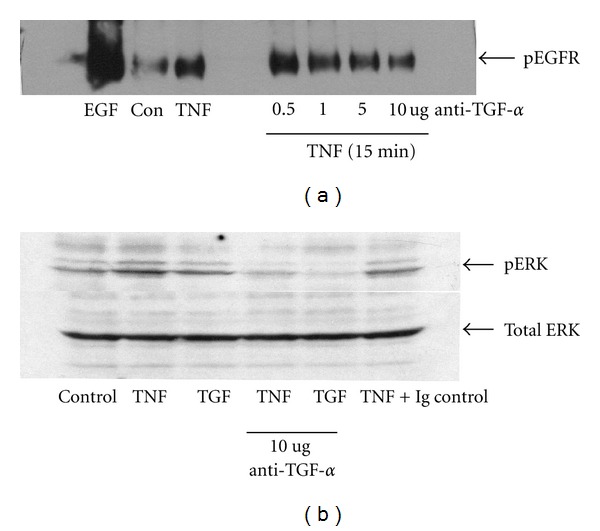
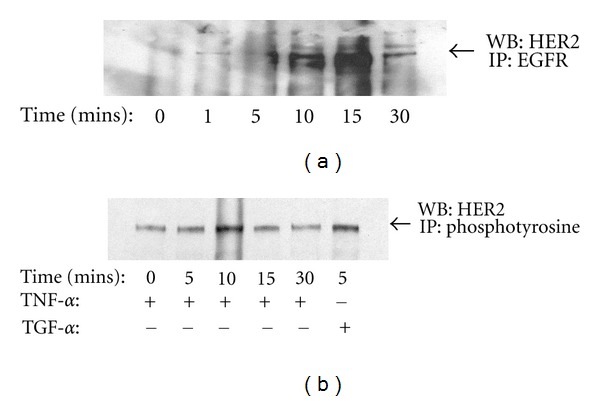
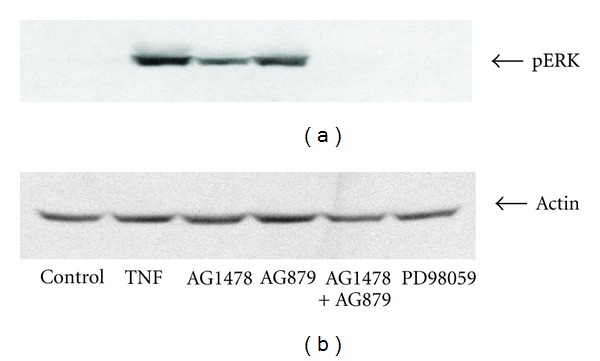
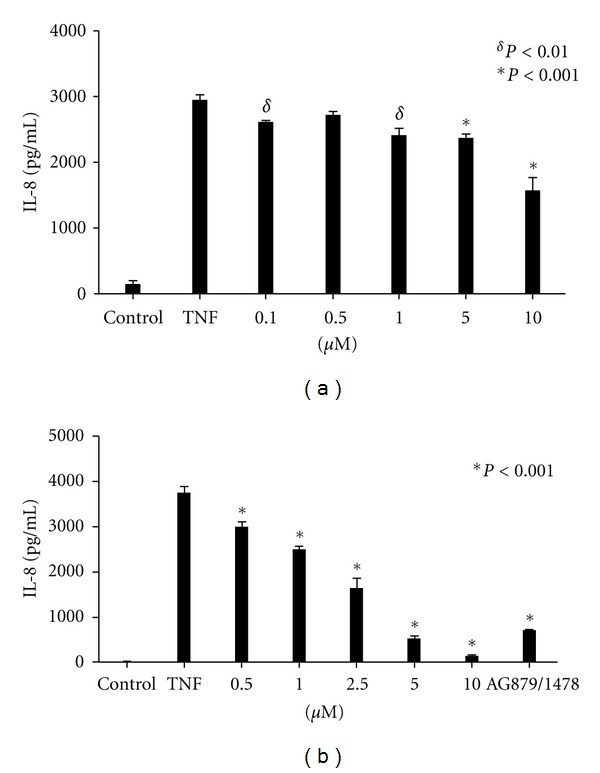
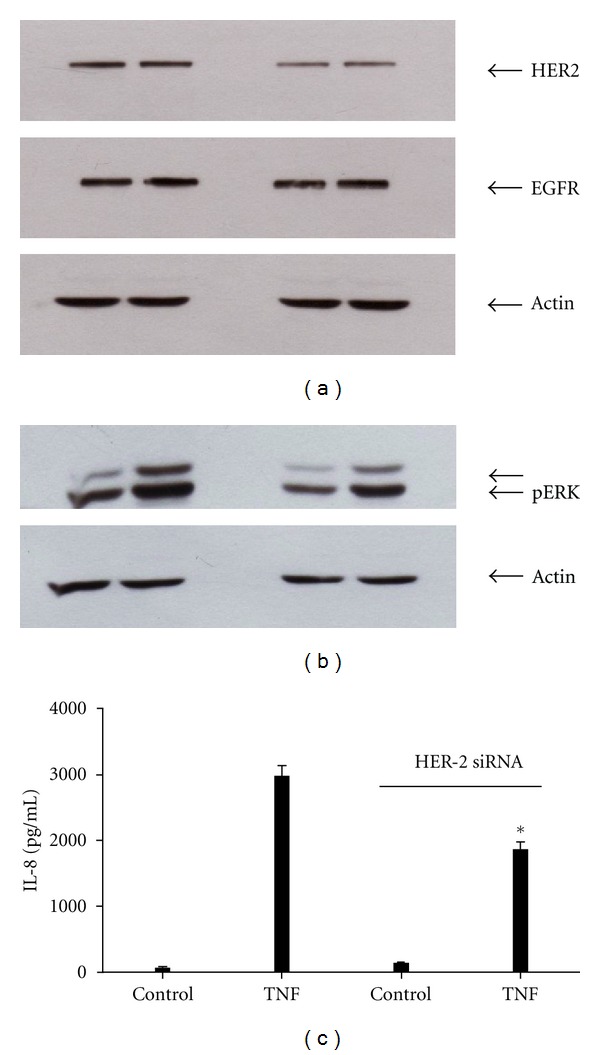
TNF-α activates multiple mitogen-activated protein kinase (MAPK) cascades in intestinal epithelial cells (IECs) leading to the secretion of interleukin 8 (IL-8), a neutrophil chemoattractant and an angiogenic factor with tumor promoting properties. As the epidermal growth factor receptor (EGFR) is a known transducer of proliferative signals and a potent activator of MAPKs, we hypothesized that the EGFR participates in TNF-dependent MAPK activation and IL-8 secretion by intestinal epithelial cells (IECs). We show that the EGFR is tyrosine-phosphorylated following treatment of IECs (HT-29 and IEC-6) with TNF-α. This requires EGFR autophosphorylation as it was blocked by the EGFR kinase inhibitor AG1478. Autophosphorylation was also inhibited by both a Src-kinase inhibitor and the metalloproteinase inhibitor batimastat. TNF treatment of IECs resulted in the accumulation of soluble TGF-α; treatment of IECs with batimastat suppressed TGF-α release and immunoneutralization of TGF-α resulted in decreased EGFR and ERK phosphorylations. TNF-α treatment of IECs resulted in an association between EGFR and HER2 and inhibition of HER2 using a specific inhibitor AG879 in combination with AG1478-suppressed TNF-α-dependent ERK phosphorylation and IL-8 release. Downregulation of HER2 via siRNA resulted in a significant decrease in ERK phosphorylation and a 50% reduction in IL-8 secretion.
Figures
Similar articles
-
Metalloprotease-dependent amphiregulin release mediates tumor necrosis factor-alpha-induced IL-8 secretion in the human airway epithelial cell line NCI-H292.Life Sci. 2006 May 22;78(26):3051-7. doi: 10.1016/j.lfs.2005.12.023. Epub 2006 Jan 19. Life Sci. 2006. PMID: 16427093
-
TNF-α and LPA promote synergistic expression of COX-2 in human colonic myofibroblasts: role of LPA-mediated transactivation of upregulated EGFR.BMC Gastroenterol. 2013 May 20;13:90. doi: 10.1186/1471-230X-13-90. BMC Gastroenterol. 2013. PMID: 23688423 Free PMC article.
-
Clostridium difficile toxin B activates the EGF receptor and the ERK/MAP kinase pathway in human colonocytes.Gastroenterology. 2005 Apr;128(4):1002-11. doi: 10.1053/j.gastro.2005.01.053. Gastroenterology. 2005. PMID: 15825081
-
Carbachol-stimulated transactivation of epidermal growth factor receptor and mitogen-activated protein kinase in T(84) cells is mediated by intracellular Ca2+, PYK-2, and p60(src).J Biol Chem. 2000 Apr 28;275(17):12619-25. doi: 10.1074/jbc.275.17.12619. J Biol Chem. 2000. PMID: 10777553
-
Transactivation of EGF receptor and ErbB2 protects intestinal epithelial cells from TNF-induced apoptosis.Proc Natl Acad Sci U S A. 2008 Aug 19;105(33):11772-7. doi: 10.1073/pnas.0801463105. Epub 2008 Aug 13. Proc Natl Acad Sci U S A. 2008. PMID: 18701712 Free PMC article.
Cited by
-
Inflammation-induced desmoglein-2 ectodomain shedding compromises the mucosal barrier.Mol Biol Cell. 2015 Sep 15;26(18):3165-77. doi: 10.1091/mbc.E15-03-0147. Epub 2015 Jul 29. Mol Biol Cell. 2015. PMID: 26224314 Free PMC article.
-
Enterococcus faecalis from healthy infants modulates inflammation through MAPK signaling pathways.PLoS One. 2014 May 15;9(5):e97523. doi: 10.1371/journal.pone.0097523. eCollection 2014. PLoS One. 2014. PMID: 24830946 Free PMC article.
-
Establishment and characterization of cetuximab resistant head and neck squamous cell carcinoma cell lines: focus on the contribution of the AP-1 transcription factor.Am J Cancer Res. 2015 May 15;5(6):1921-38. eCollection 2015. Am J Cancer Res. 2015. PMID: 26269754 Free PMC article.
-
Inhibition of cell invasion and migration by targeting matrix metalloproteinase-9 expression via sirtuin 6 silencing in human breast cancer cells.Sci Rep. 2022 Jul 15;12(1):12125. doi: 10.1038/s41598-022-16405-x. Sci Rep. 2022. PMID: 35840633 Free PMC article.
-
Enterococcus faecium HDRsEf1 Protects the Intestinal Epithelium and Attenuates ETEC-Induced IL-8 Secretion in Enterocytes.Mediators Inflamm. 2016;2016:7474306. doi: 10.1155/2016/7474306. Epub 2016 Nov 7. Mediators Inflamm. 2016. PMID: 27890970 Free PMC article.
References
-
- Sartor RB, Muehlbauer M. Microbial host interactions in IBD: implications for pathogenesis and therapy. Current Gastroenterology Reports. 2007;9(6):497–507. - PubMed
-
- Wilson JAP. Tumor necrosis factor α and colitis-associated colon cancer. The New England Journal of Medicine. 2008;358(25):2733–2734. - PubMed
-
- Balkwill F, Coussens LM. Cancer: an inflammatory link. Nature. 2004;431(7007):405–406. - PubMed
-
- Bates RC, Deleo MJ, Mercurio AM. The epithelial-mesenchymal transition of colon carcinoma involves expression of IL-8 and CXCR-1-mediated chemotaxis. Experimental Cell Research. 2004;299(2):315–324. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
