

OSU-03012 interacts with lapatinib to kill brain cancer cells
- PMID: 22990204
- PMCID: PMC3542242
- DOI: 10.4161/cbt.22275
OSU-03012 interacts with lapatinib to kill brain cancer cells
Abstract
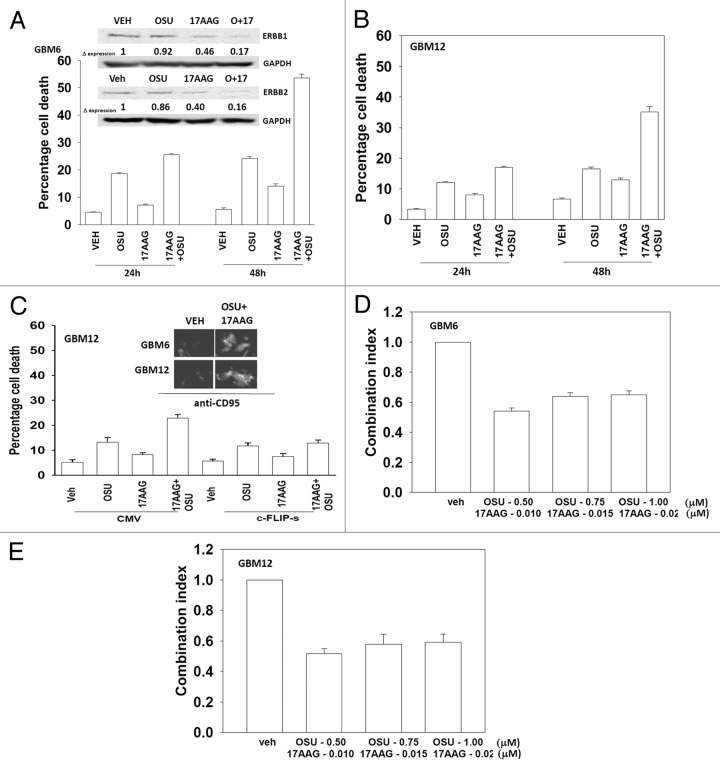
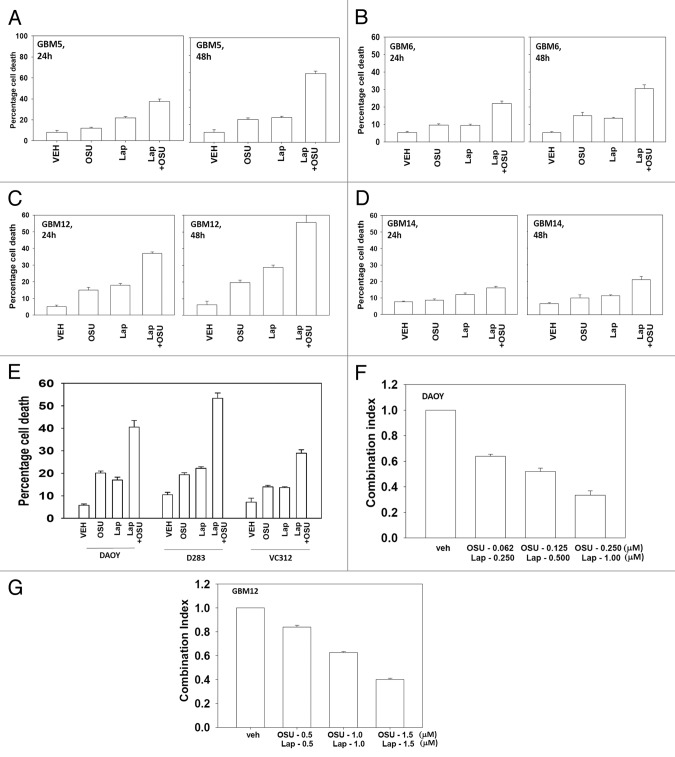
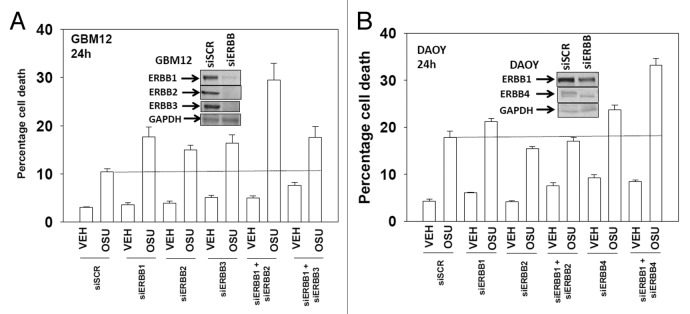
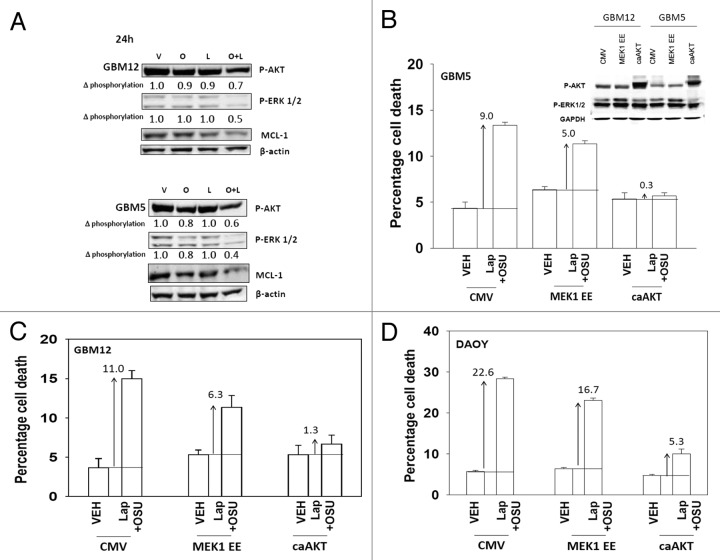
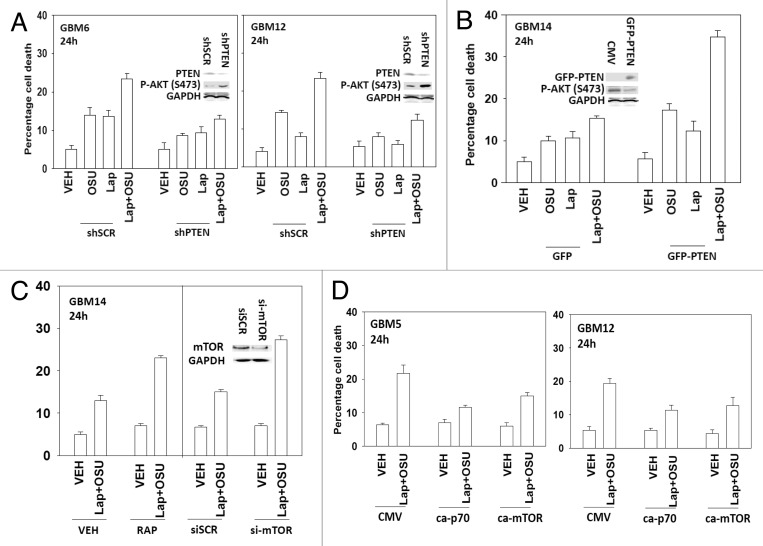
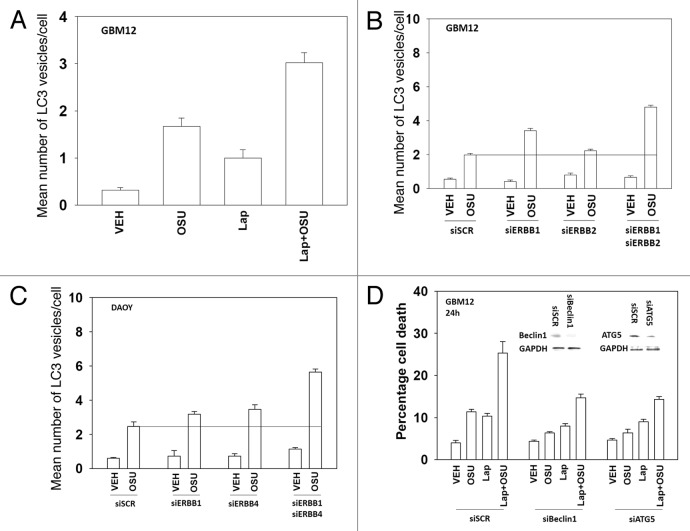
We have further defined mechanism(s) by which the drug OSU-03012 (OSU) kills brain cancer cells. OSU toxicity was enhanced by the HSP90 inhibitor 17-N-Allylamino-17-demethoxygeldanamycin (17AAG) that correlated with reduced expression of ERBB1 and ERBB2. Inhibition of the extrinsic apoptosis pathway blocked the interaction between 17AAG and OSU. OSU toxicity was enhanced by the inhibitor of ERBB1/2/4, lapatinib. Knock down of ERBB1/2/4 in a cell line specific fashion promoted OSU toxicity. Combined exposure of cells to lapatinib and OSU resulted in reduced AKT and ERK1/2 activity; expression of activated forms of AKT and to a lesser extent MEK1 protected cells from the lethal effects of the drug combination. Knock down of PTEN suppressed, and expression of PTEN enhanced, the lethal interaction between OSU and lapatinib. Downstream of PTEN, inhibition of mTOR recapitulated the effects of lapatinib. Knock down of CD95, NOXA, PUMA, BIK or AIF, suppressed lapatinib and OSU toxicity. Knock down of MCL-1 enhanced, and overexpression of MCL-1 suppressed, drug combination lethality. Lapatinib and OSU interacted in vivo to suppress the growth of established tumors. Collectively our data argue that the inhibition of ERBB receptor function represents a useful way to enhance OSU lethality in brain tumor cells.
Figures


References
-
- Hawkey CJ, Fortun PJ. Cyclooxygenase-2 inhibitors. Curr Opin Gastroenterol. 2005;21:660–4. - PubMed
-
- Kiefer W, Dannhardt G. Novel insights and therapeutical applications in the field of inhibitors of COX-2. Curr Med Chem. 2004;11:3147–61. - PubMed
-
- Kang SG, Kim JS, Park K, Kim JS, Groves MD, Nam DH. Combination celecoxib and temozolomide in C6 rat glioma orthotopic model. Oncol Rep. 2006;15:7–13. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous