

Streamlining and core genome conservation among highly divergent members of the SAR11 clade
- PMID: 22991429
- PMCID: PMC3448164
- DOI: 10.1128/mBio.00252-12
Streamlining and core genome conservation among highly divergent members of the SAR11 clade
Abstract
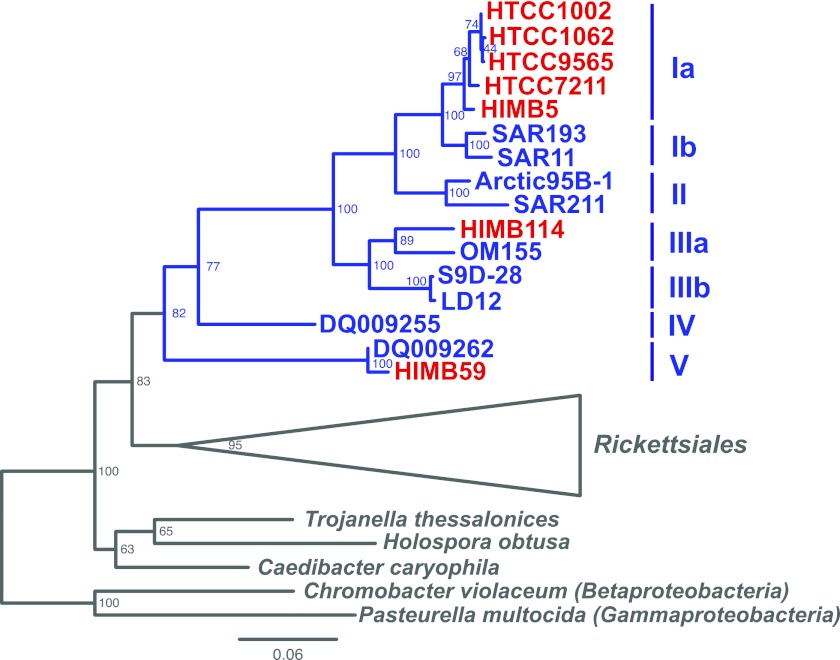
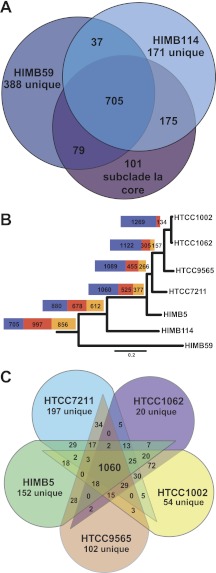
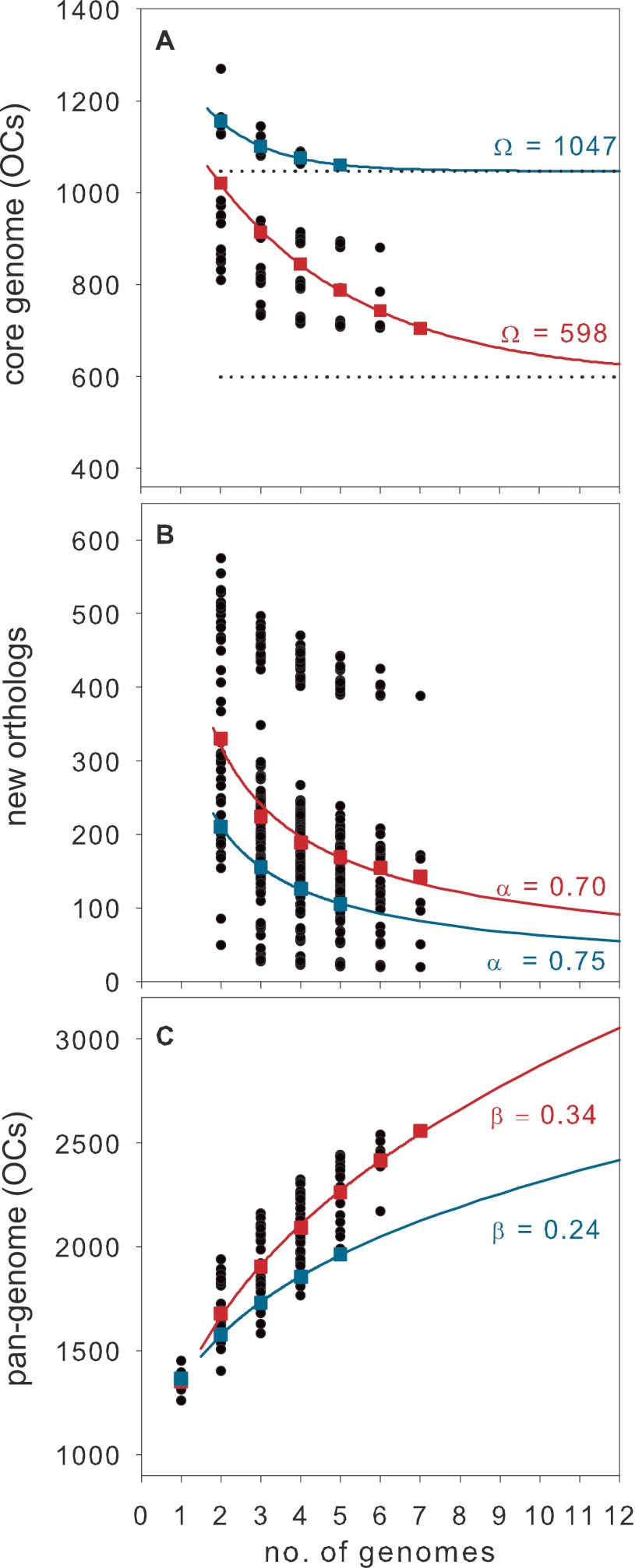
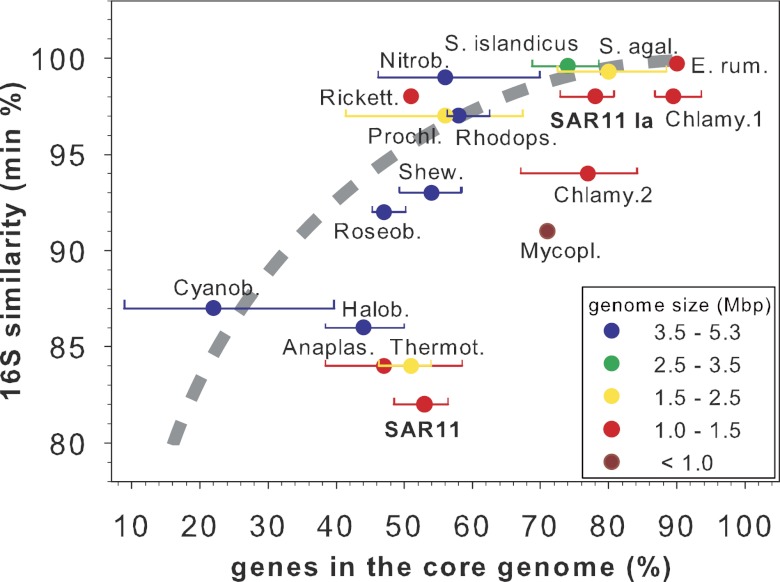
SAR11 is an ancient and diverse clade of heterotrophic bacteria that are abundant throughout the world's oceans, where they play a major role in the ocean carbon cycle. Correlations between the phylogenetic branching order and spatiotemporal patterns in cell distributions from planktonic ocean environments indicate that SAR11 has evolved into perhaps a dozen or more specialized ecotypes that span evolutionary distances equivalent to a bacterial order. We isolated and sequenced genomes from diverse SAR11 cultures that represent three major lineages and encompass the full breadth of the clade. The new data expand observations about genome evolution and gene content that previously had been restricted to the SAR11 Ia subclade, providing a much broader perspective on the clade's origins, evolution, and ecology. We found small genomes throughout the clade and a very high proportion of core genome genes (48 to 56%), indicating that small genome size is probably an ancestral characteristic. In their level of core genome conservation, the members of SAR11 are outliers, the most conserved free-living bacteria known. Shared features of the clade include low GC content, high gene synteny, a large hypervariable region bounded by rRNA genes, and low numbers of paralogs. Variation among the genomes included genes for phosphorus metabolism, glycolysis, and C1 metabolism, suggesting that adaptive specialization in nutrient resource utilization is important to niche partitioning and ecotype divergence within the clade. These data provide support for the conclusion that streamlining selection for efficient cell replication in the planktonic habitat has occurred throughout the evolution and diversification of this clade. IMPORTANCE The SAR11 clade is the most abundant group of marine microorganisms worldwide, making them key players in the global carbon cycle. Growing knowledge about their biochemistry and metabolism is leading to a more mechanistic understanding of organic carbon oxidation and sequestration in the oceans. The discovery of small genomes in SAR11 provided crucial support for the theory that streamlining selection can drive genome reduction in low-nutrient environments. Study of isolates in culture revealed atypical organic nutrient requirements that can be attributed to genome reduction, such as conditional auxotrophy for glycine and its precursors, a requirement for reduced sulfur compounds, and evidence for widespread cycling of C1 compounds in marine environments. However, understanding the genetic variation and distribution of such pathways and characteristics like streamlining throughout the group has required the isolation and genome sequencing of diverse SAR11 representatives, an analysis of which we provide here.
Figures
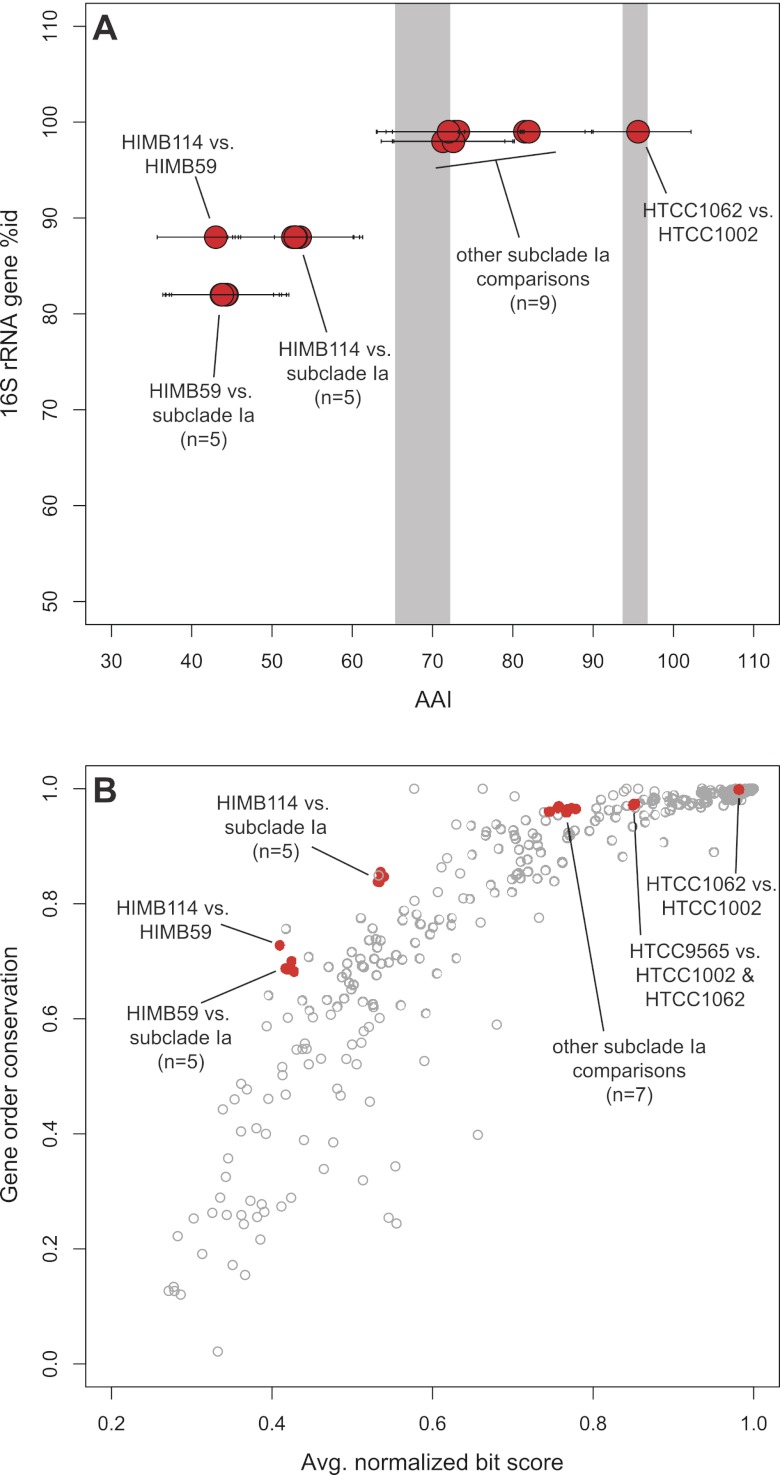
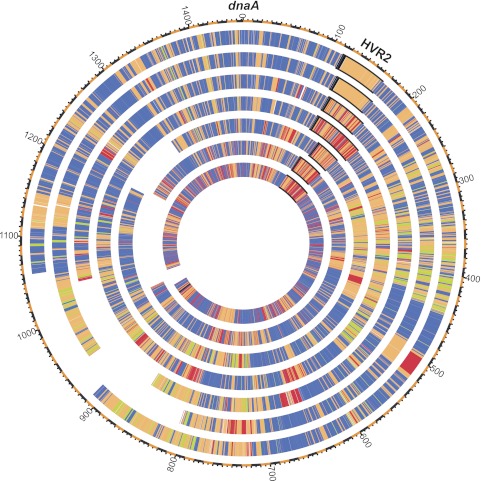
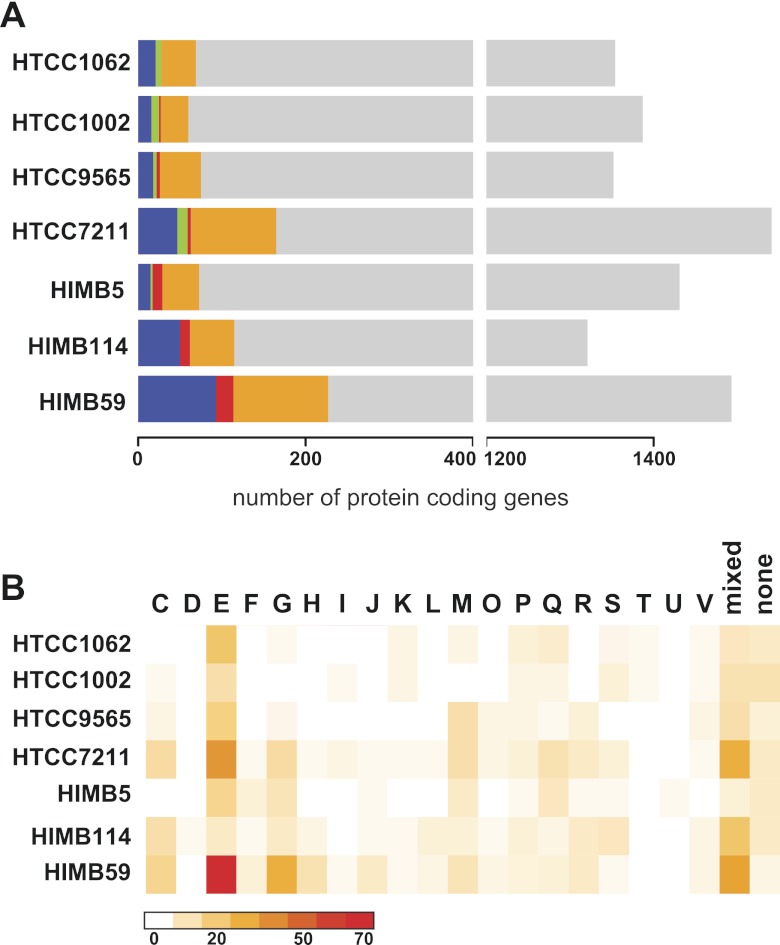
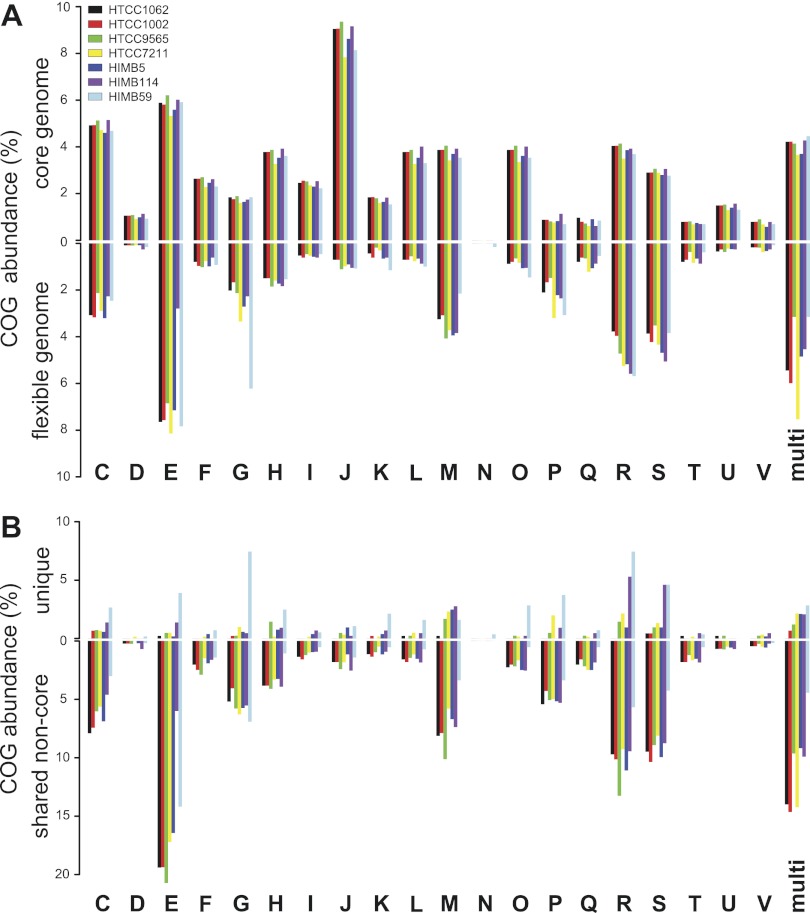
References
-
- Morris RM, et al. 2002. SAR11 clade dominates ocean surface bacterioplankton communities. Nature 420:806–810 - PubMed
-
- Schattenhofer M, et al. 2009. Latitudinal distribution of prokaryotic picoplankton populations in the Atlantic Ocean. Environ. Microbiol. 11:2078–2093 - PubMed
-
- Rocap G, et al. 2003. Genome divergence in two Prochlorococcus ecotypes reflects oceanic niche differentiation. Nature 424:1042–1047 - PubMed
-
- Rappé MS, Connon SA, Vergin KL, Giovannoni SJ. 2002. Cultivation of the ubiquitous SAR11 marine bacterioplankton clade. Nature 418:630–633 - PubMed
-
- Giovannoni SJ, et al. 2005. Genome streamlining in a cosmopolitan oceanic bacterium. Science 309:1242–1245 - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Miscellaneous
