

Assessing the range of kinase autoinhibition mechanisms in the insulin receptor family
- PMID: 22992069
- PMCID: PMC3492919
- DOI: 10.1042/BJ20121365
Assessing the range of kinase autoinhibition mechanisms in the insulin receptor family
Abstract
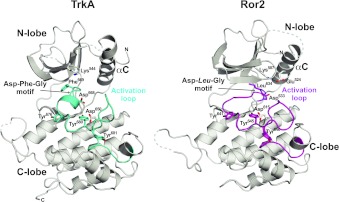
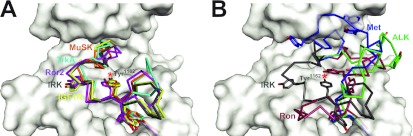
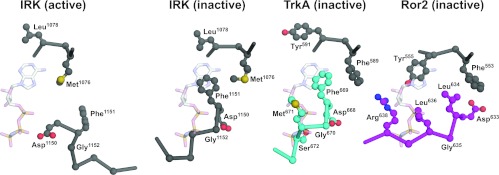
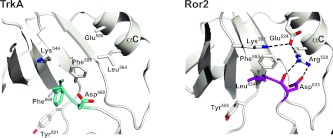
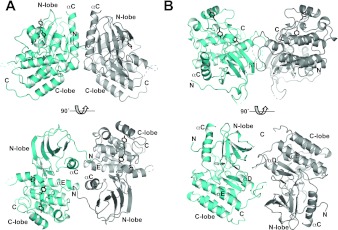
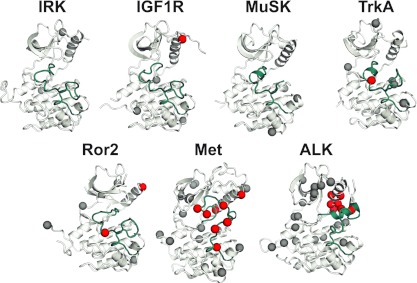
To investigate the range of autoinhibitory mechanisms used by TKDs (tyrosine kinase domains) from the insulin receptor family of RTKs (receptor tyrosine kinases), we determined crystal structures of TKDs from TrkA (tropomyosin receptor kinase A, a nerve growth factor receptor) and Ror2 (receptor tyrosine kinase-like orphan receptor 2, an unconventional Wnt receptor). TrkA autoinhibition closely resembles that seen for the insulin receptor, relying on projection of an activation loop tyrosine residue into the substrate-binding site and occlusion of the ATP-binding site by the activation loop. Ror2 employs similar mechanisms, but the unusual replacement of the phenylalanine residue in its Asp-Phe-Gly motif with leucine necessitates occlusion of the ATP-binding site by other means. The unusual Asp-Leu-Gly motif in Ror2 is displaced compared with other inactive kinases, allowing the activation loop to interact directly with the TKD's αC helix, in another mode of autoinhibition that is characteristic of the other extreme of this receptor family: ALK (anaplastic lymphoma kinase) and Met. These findings provide insight into the expected range of activating mutations in these TKDs in cancer. We also describe symmetrical dimers of the inactive TrkA TKD resembling those found in other RTKs, possibly reflecting an arrangement of kinase domains in a pre-formed TrkA dimer.
Figures
References
-
- Dancey J. E., Bedard P. L., Onetto N., Hudson T. J. The genetic basis for cancer treatment decisions. Cell. 2012;148:409–420. - PubMed
-
- Yauch R. L., Settleman J. Recent advances in pathway-targeted cancer drug therapies emerging from cancer genome analysis. Curr. Opin. Genet. Dev. 2012;22:45–49. - PubMed
-
- Zhang X., Gureasko J., Shen K., Cole P. A., Kuriyan J. An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell. 2006;125:1137–1149. - PubMed
-
- Sharma S. V., Bell D. W., Settleman J., Haber D. A. Epidermal growth factor receptor mutations in lung cancer. Nat. Rev. Cancer. 2007;7:169–181. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous