

The T1048I mutation in ATP7A gene causes an unusual Menkes disease presentation
- PMID: 22992316
- PMCID: PMC3489546
- DOI: 10.1186/1471-2431-12-150
The T1048I mutation in ATP7A gene causes an unusual Menkes disease presentation
Abstract
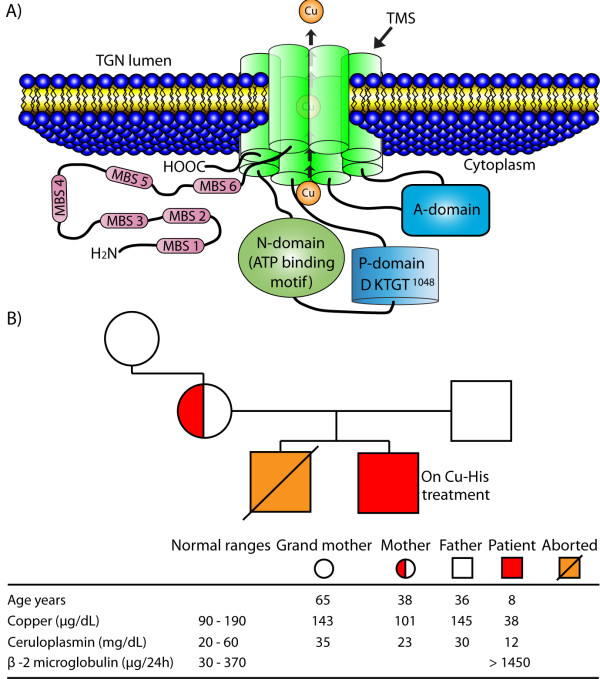
Background: The ATP7A gene encodes the ATP7A protein, which is a trans-Golgi network copper transporter expressed in the brain and other organs. Mutations in this gene cause disorders of copper metabolism, such as Menkes disease. Here we describe the novel and unusual mutation (p.T1048I) in the ATP7A gene of a child with Menkes disease. The mutation affects a conserved DKTGT1048 phosphorylation motif that is involved in the catalytic activity of ATP7A. We also describe the clinical course and the response to copper treatment in this patient.
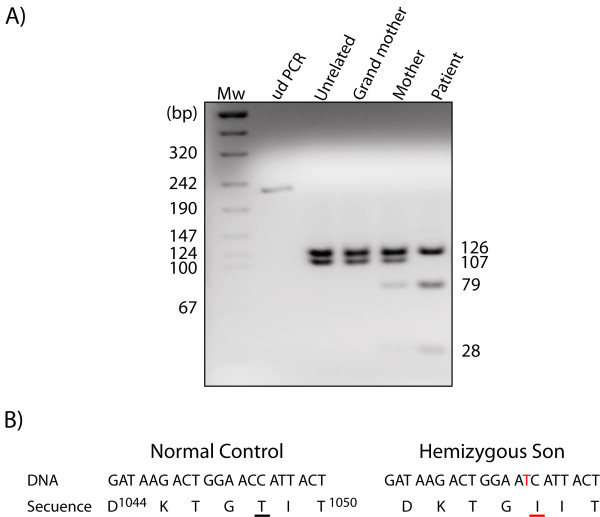
Case presentation: An 11-month-old male Caucasian infant was studied because of hypotonia, ataxia and global developmental delay. The patient presented low levels of serum copper and ceruloplasmin, and was shown to be hemizygous for the p.T1048I mutation in ATP7A. The diagnosis was confirmed when the patient was 18 months old, and treatment with copper-histidinate (Cu-His) was started immediately. The patient showed some neurological improvement and he is currently 8 years old. Because the p.T1048I mutation affects its catalytic site, we expected a complete loss of functional ATP7A and a classical Menkes disease presentation. However, the clinical course of the patient was mild, and he responded to Cu-His treatment, which suggests that this mutation leads to partial conservation of the activity of ATP7A.
Conclusion: This case emphasizes the important correlation between genotype and phenotype in patients with Menkes disease. The prognosis in Menkes disease is associated with early detection, early initiation of treatment and with the preservation of some ATP7A activity, which is necessary for Cu-His treatment response. The description of this new mutation and the response of the patient to Cu-His treatment will contribute to the growing body of knowledge about treatment response in Menkes disease.
Figures
Similar articles
-
In utero copper treatment for Menkes disease associated with a severe ATP7A mutation.Mol Genet Metab. 2012 Sep;107(1-2):222-8. doi: 10.1016/j.ymgme.2012.05.008. Epub 2012 May 18. Mol Genet Metab. 2012. PMID: 22695177 Free PMC article.
-
A Truncating De Novo Point Mutation in a Young Infant with Severe Menkes Disease.Pediatr Neonatol. 2017 Feb;58(1):89-92. doi: 10.1016/j.pedneo.2014.05.008. Epub 2014 Nov 14. Pediatr Neonatol. 2017. PMID: 25771438
-
Copper-histidine therapy in an infant with novel splice-site variant in the ATP7A gene of Menkes disease: the first experience in South East Asia and literature review.BMJ Case Rep. 2022 Apr 7;15(4):e247937. doi: 10.1136/bcr-2021-247937. BMJ Case Rep. 2022. PMID: 35393273 Free PMC article. Review.
-
Novel mutations and clinical outcomes of copper-histidine therapy in Menkes disease patients.Metab Brain Dis. 2015 Feb;30(1):75-81. doi: 10.1007/s11011-014-9569-5. Epub 2014 Jun 13. Metab Brain Dis. 2015. PMID: 24919650
-
[From gene to disease; Menkes disease: copper deficiency due to an ATP7A-gene defect].Ned Tijdschr Geneeskd. 2007 Oct 13;151(41):2266-70. Ned Tijdschr Geneeskd. 2007. PMID: 17987894 Review. Dutch.
Cited by
-
The Affymetrix DMET Plus platform reveals unique distribution of ADME-related variants in ethnic Arabs.Dis Markers. 2015;2015:542543. doi: 10.1155/2015/542543. Epub 2015 Feb 22. Dis Markers. 2015. PMID: 25802476 Free PMC article.
-
ATP7A Clinical Genetics Resource - A comprehensive clinically annotated database and resource for genetic variants in ATP7A gene.Comput Struct Biotechnol J. 2020 Sep 2;18:2347-2356. doi: 10.1016/j.csbj.2020.08.021. eCollection 2020. Comput Struct Biotechnol J. 2020. PMID: 32994893 Free PMC article.
-
Identification of novel ATP7A mutations and prenatal diagnosis in Chinese patients with Menkes disease.Metab Brain Dis. 2017 Aug;32(4):1123-1131. doi: 10.1007/s11011-017-9985-4. Epub 2017 Apr 10. Metab Brain Dis. 2017. PMID: 28397151
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
