

Crystallization, dehydration and experimental phasing of WbdD, a bifunctional kinase and methyltransferase from Escherichia coli O9a
- PMID: 22993091
- PMCID: PMC3447403
- DOI: 10.1107/S0907444912029599
Crystallization, dehydration and experimental phasing of WbdD, a bifunctional kinase and methyltransferase from Escherichia coli O9a
Abstract
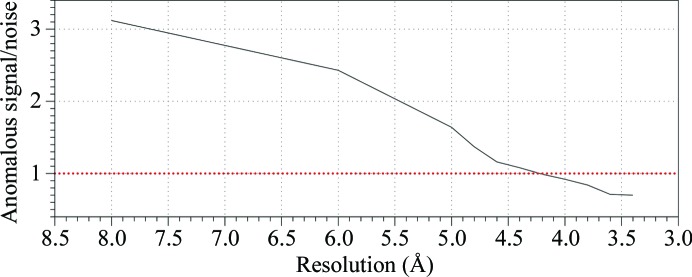
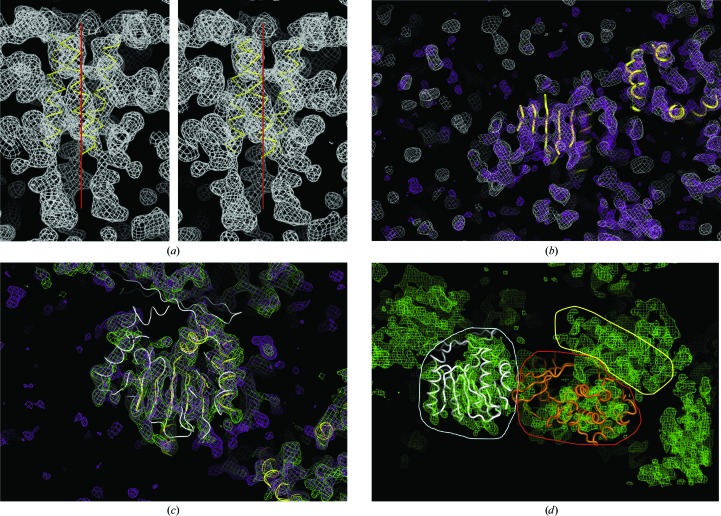
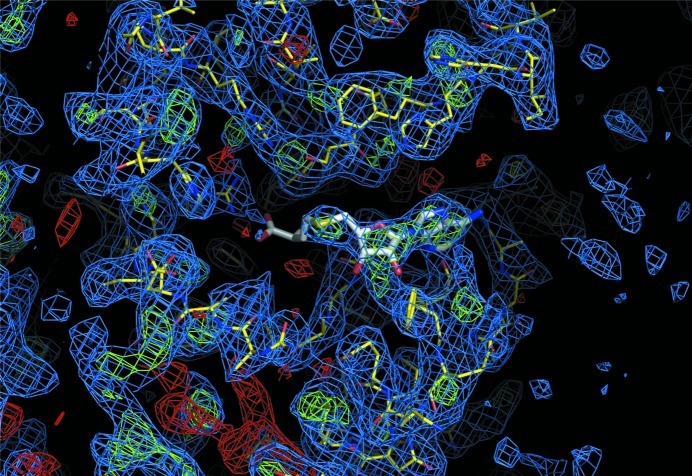
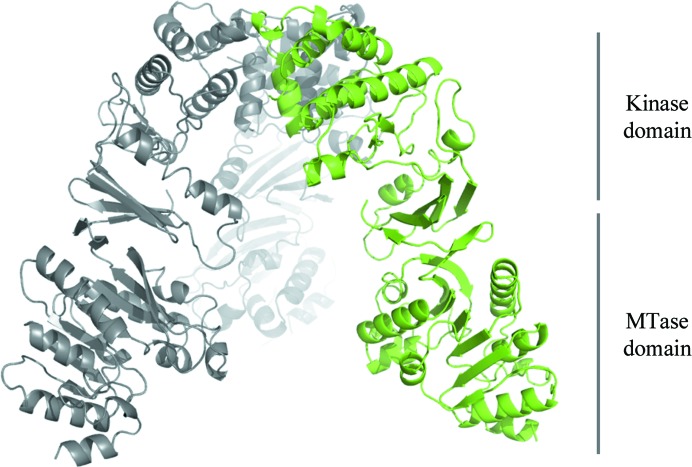
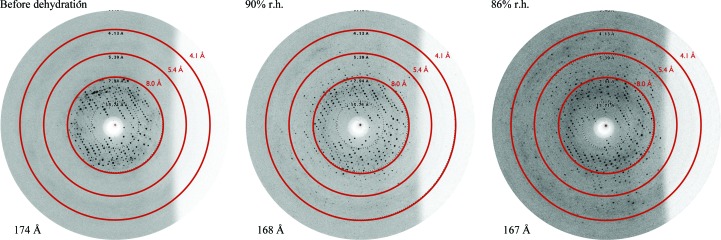
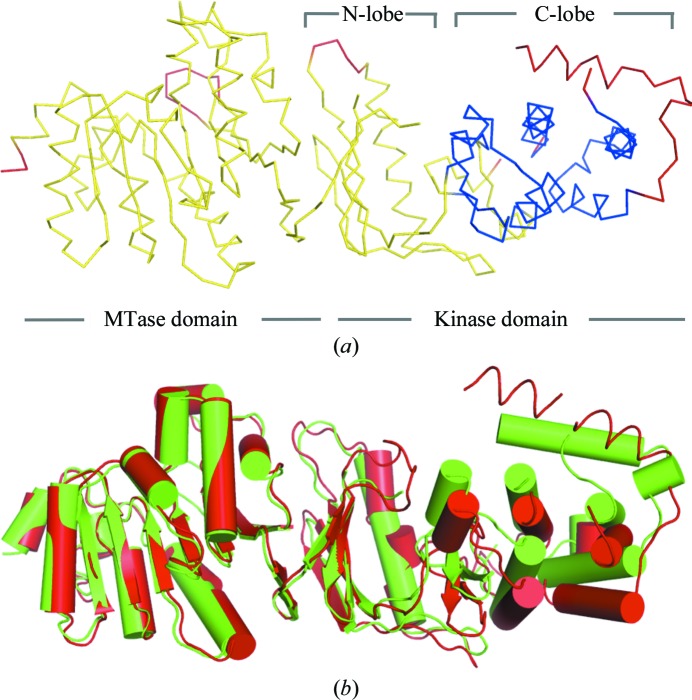
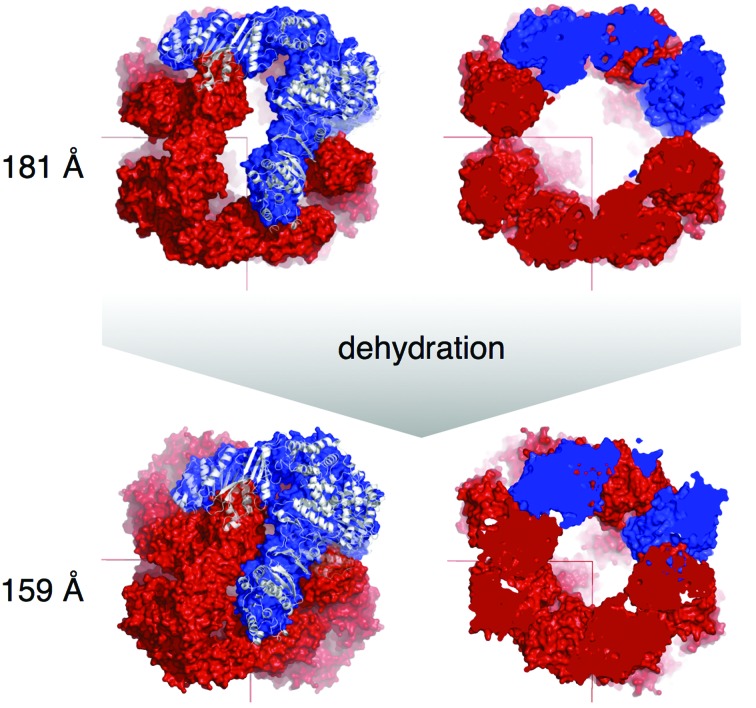
WbdD is a bifunctional kinase/methyltransferase that is responsible for regulation of lipopolysaccharide O antigen polysaccharide chain length in Escherichia coli serotype O9a. Solving the crystal structure of this protein proved to be a challenge because the available crystals belonging to space group I23 only diffracted to low resolution (>95% of the crystals diffracted to resolution lower than 4 Å and most only to 8 Å) and were non-isomorphous, with changes in unit-cell dimensions of greater than 10%. Data from a serendipitously found single native crystal that diffracted to 3.0 Å resolution were non-isomorphous with a lower (3.5 Å) resolution selenomethionine data set. Here, a strategy for improving poor (3.5 Å resolution) initial phases by density modification and cross-crystal averaging with an additional 4.2 Å resolution data set to build a crude model of WbdD is desribed. Using this crude model as a mask to cut out the 3.5 Å resolution electron density yielded a successful molecular-replacement solution of the 3.0 Å resolution data set. The resulting map was used to build a complete model of WbdD. The hydration status of individual crystals appears to underpin the variable diffraction quality of WbdD crystals. After the initial structure had been solved, methods to control the hydration status of WbdD were developed and it was thus possible to routinely obtain high-resolution diffraction (to better than 2.5 Å resolution). This novel and facile crystal-dehydration protocol may be useful for similar challenging situations.
Figures

References
-
- Adams, P. D., Grosse-Kunstleve, R. W., Hung, L.-W., Ioerger, T. R., McCoy, A. J., Moriarty, N. W., Read, R. J., Sacchettini, J. C., Sauter, N. K. & Terwilliger, T. C. (2002). Acta Cryst. D58, 1948–1954. - PubMed
-
- Clarke, B. R., Cuthbertson, L. & Whitfield, C. (2004). J. Biol. Chem. 279, 35709–35718. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
