

Inference on population histories by approximating infinite alleles diffusion
- PMID: 22993237
- PMCID: PMC3548313
- DOI: 10.1093/molbev/mss227
Inference on population histories by approximating infinite alleles diffusion
Abstract
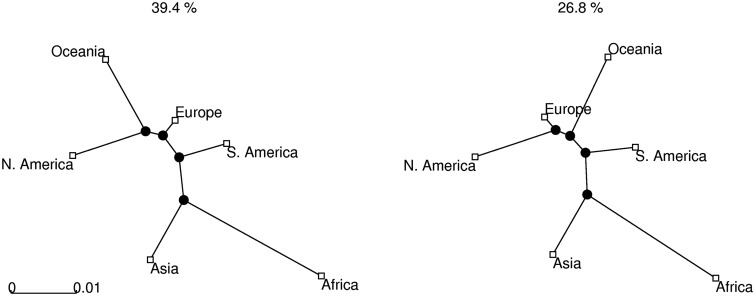
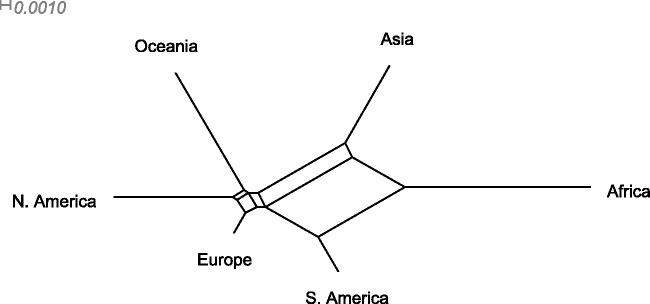
Reconstruction of the past is an important task of evolutionary biology. It takes place at different points in a hierarchy of molecular variation, including genes, individuals, populations, and species. Statistical inference about population histories has recently received considerable attention, following the development of computational tools to provide tractable approaches to this very challenging problem. Here, we introduce a likelihood-based approach which generalizes a recently developed model for random fluctuations in allele frequencies based on an approximation to the neutral Wright-Fisher diffusion. Our new framework approximates the infinite alleles Wright-Fisher model and uses an implementation with an adaptive Markov chain Monte Carlo algorithm. The method is especially well suited to data sets harboring large population samples and relatively few loci for which other likelihood-based models are currently computationally intractable. Using our model, we reconstruct the global population history of a major human pathogen, Streptococcus pneumoniae. The results illustrate the potential to reach important biological insights to an evolutionary process by a population genetics approach, which can appropriately accommodate very large population samples.
Figures
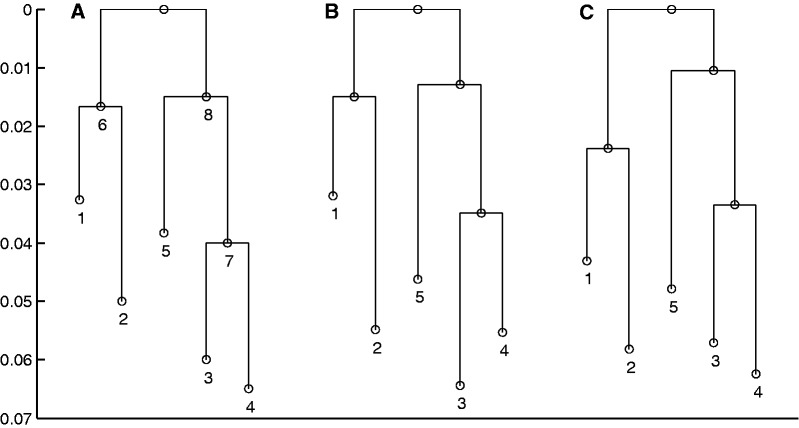
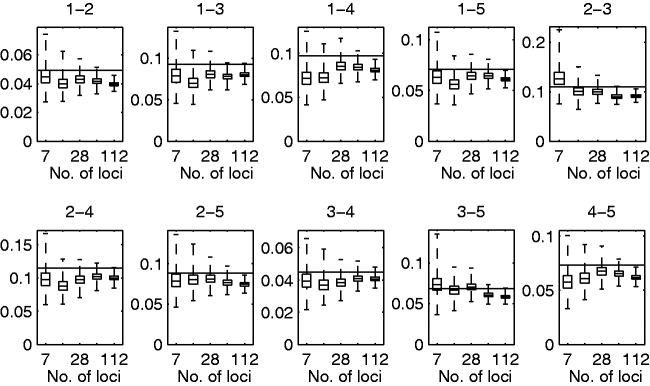
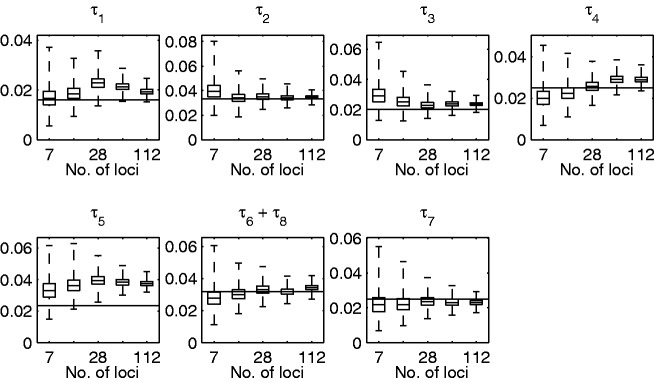
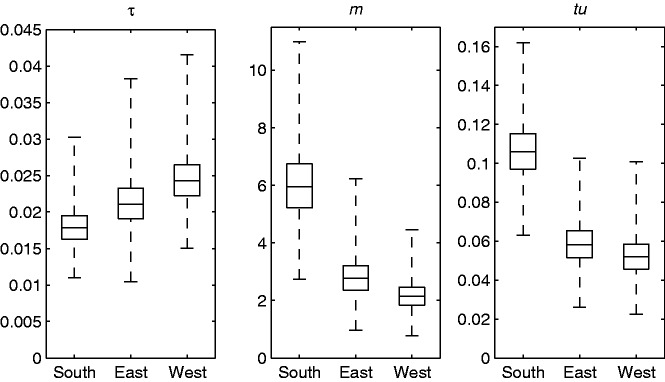
References
-
- Balding DJ, Nichols RA. A method for quantifying differentiation between populations at multi-allelic loci and its implications for investigating identity and paternity. Genetica. 1995;96:3–12. - PubMed
-
- Bryant D, Moulton V. Neighbor-net: an agglomerative method for the construction of phylogenetic networks. Mol Biol Evol. 2004;21:255–265. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
