

Endogenous IRAK-M attenuates postinfarction remodeling through effects on macrophages and fibroblasts
- PMID: 22995519
- PMCID: PMC3510666
- DOI: 10.1161/ATVBAHA.112.300310
Endogenous IRAK-M attenuates postinfarction remodeling through effects on macrophages and fibroblasts
Abstract
Objective: Effective postinfarction repair requires timely suppression of innate immune signals to prevent the catastrophic consequences of uncontrolled inflammation on cardiac geometry and function. In macrophages, interleukin-1 receptor-associated kinase (IRAK)-M acts as a functional decoy preventing uncontrolled toll-like receptor /interleukin-1-mediated responses. Our study investigates the role of IRAK-M as a negative regulator of the postinfarction inflammatory response and as a modulator of cardiac remodeling.
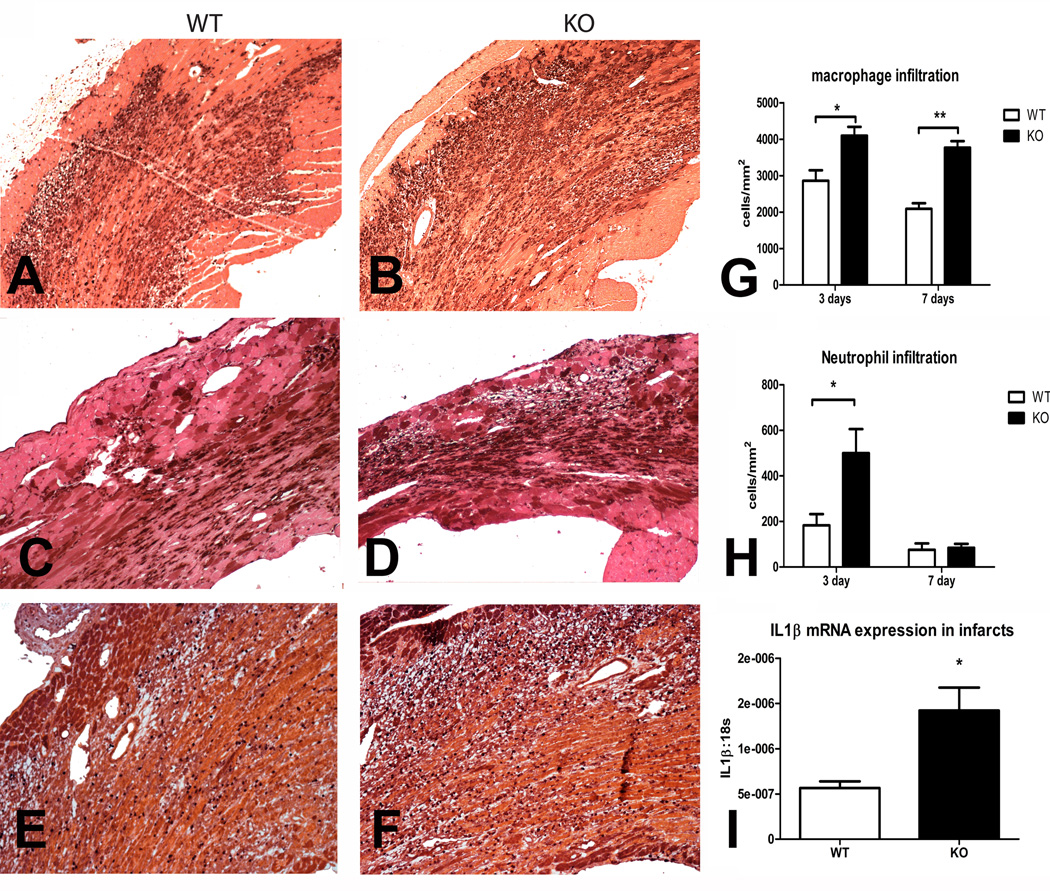
Methods and results: In wild-type mouse infarcts IRAK-M was upregulated in infiltrating macrophages and fibroblasts exhibiting a biphasic response. When compared with wild-type animals, infarcted IRAK-M(-/-) mice had enhanced adverse remodeling and worse systolic dysfunction; however, acute infarct size was comparable between groups. Adverse remodeling in IRAK-M(-/-) animals was associated with enhanced myocardial inflammation and protease activation. The protective actions of IRAK-M involved phenotypic modulation of macrophages and fibroblasts. IRAK-M(-/-) infarcts showed increased infiltration with proinflammatory CD11b+/Ly6C(hi) monocytes; leukocytes harvested from IRAK-M-null infarcts exhibited accentuated cytokine expression. In vitro, IRAK-M expression was upregulated in cytokine-stimulated murine cardiac fibroblasts and suppressed their matrix-degrading properties without affecting their inflammatory activity.
Conclusions: Endogenous IRAK-M attenuates adverse postinfarction remodeling suppressing leukocyte inflammatory activity, while inhibiting fibroblast-mediated matrix degradation.
Figures





References
-
- Nathan C, Ding A. Nonresolving inflammation. Cell. 2010;140:871–882. - PubMed
-
- Maskrey BH, Megson IL, Whitfield PD, Rossi AG. Mechanisms of resolution of inflammation: a focus on cardiovascular disease. Arterioscler Thromb Vasc Biol. 2011;31:1001–1006. - PubMed
-
- Pfeffer MA, Pfeffer JM. Ventricular enlargement and reduced survival after myocardial infarction. Circulation. 1987;75:IV93–IV97. - PubMed
-
- Holmes JW, Borg TK, Covell JW. Structure and mechanics of healing myocardial infarcts. Annu Rev Biomed Eng. 2005;7:223–253. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
