

Pemphigus autoantibodies generated through somatic mutations target the desmoglein-3 cis-interface
- PMID: 22996451
- PMCID: PMC3461925
- DOI: 10.1172/JCI64413
Pemphigus autoantibodies generated through somatic mutations target the desmoglein-3 cis-interface
Abstract
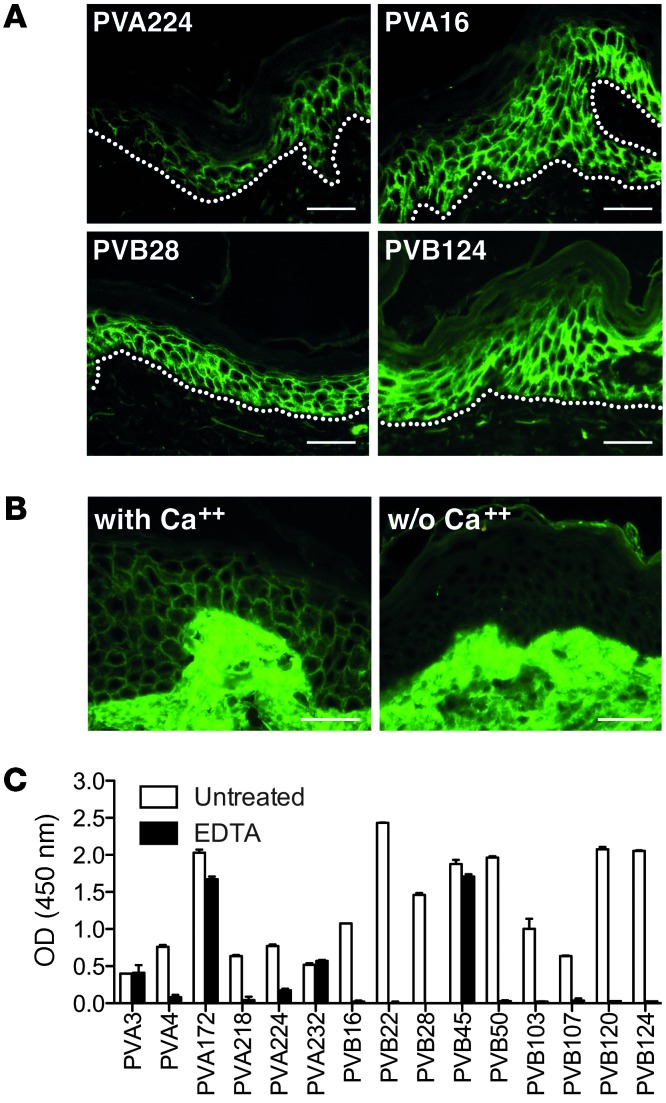
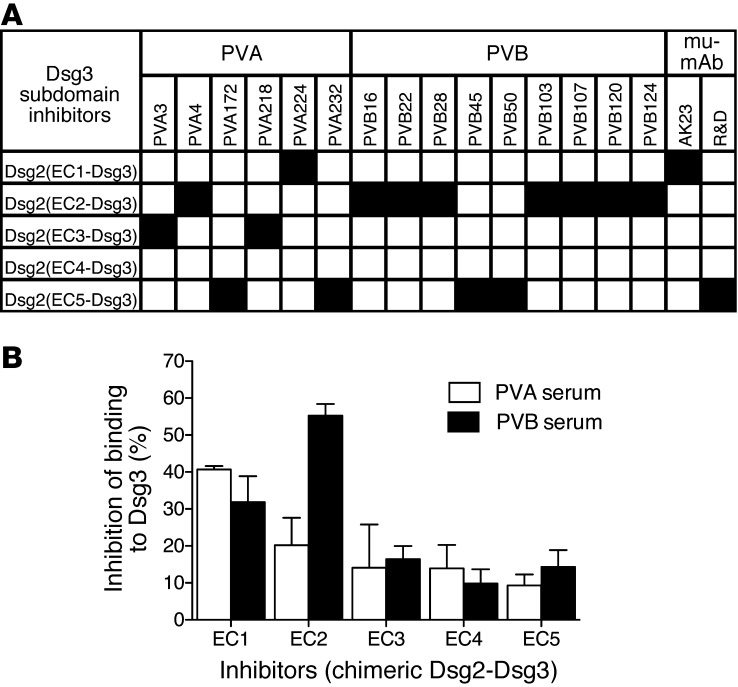
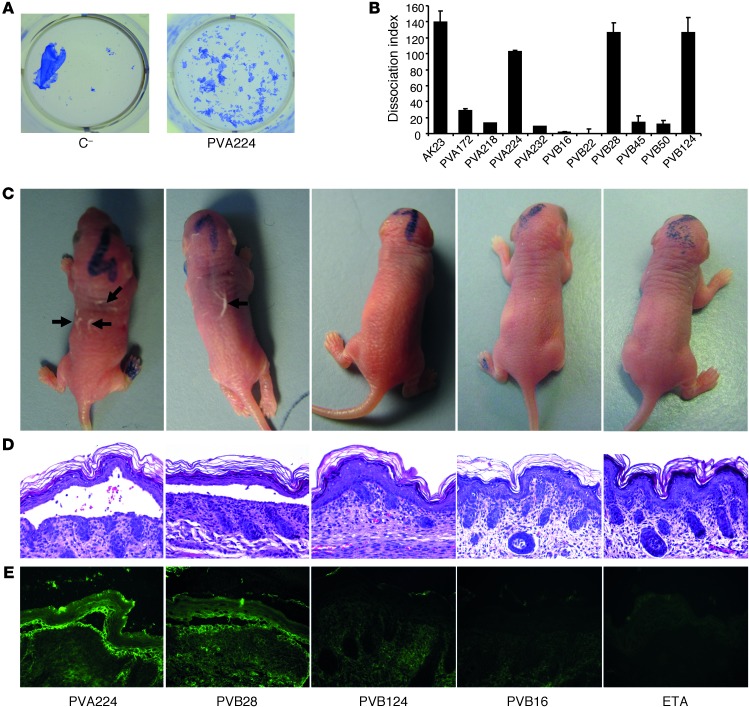
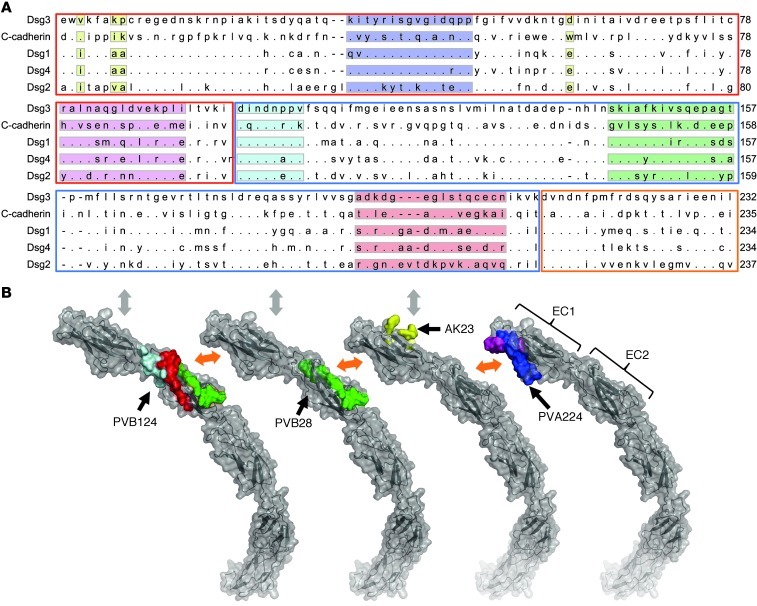
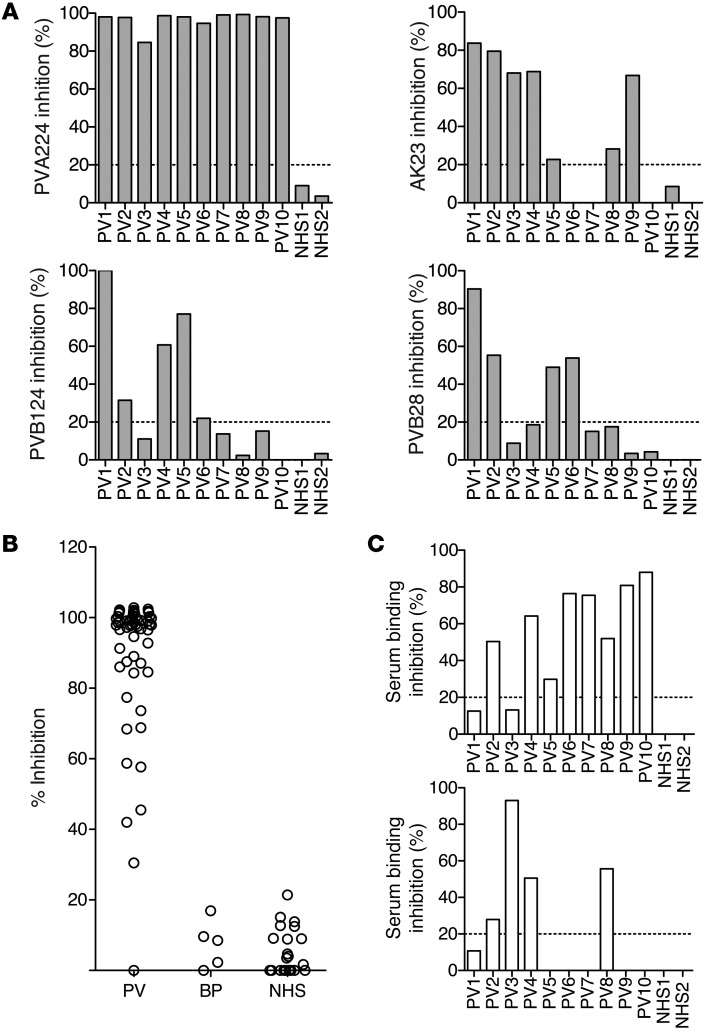
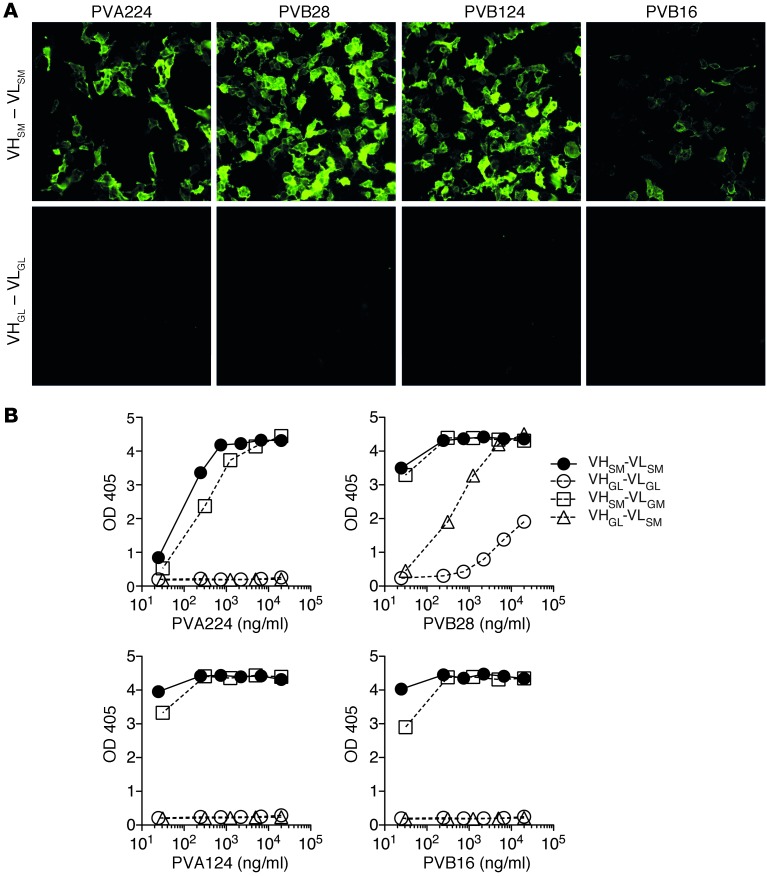
Pemphigus vulgaris (PV) is an autoimmune blistering disease of skin and mucous membranes caused by autoantibodies to the desmoglein (DSG) family proteins DSG3 and DSG1, leading to loss of keratinocyte cell adhesion. To learn more about pathogenic PV autoantibodies, we isolated 15 IgG antibodies specific for DSG3 from 2 PV patients. Three antibodies disrupted keratinocyte monolayers in vitro, and 2 were pathogenic in a passive transfer model in neonatal mice. The epitopes recognized by the pathogenic antibodies were mapped to the DSG3 extracellular 1 (EC1) and EC2 subdomains, regions involved in cis-adhesive interactions. Using a site-specific serological assay, we found that the cis-adhesive interface on EC1 recognized by the pathogenic antibody PVA224 is the primary target of the autoantibodies present in the serum of PV patients. The autoantibodies isolated used different heavy- and light-chain variable region genes and carried high levels of somatic mutations in complementary-determining regions, consistent with antigenic selection. Remarkably, binding to DSG3 was lost when somatic mutations were reverted to the germline sequence. These findings identify the cis-adhesive interface of DSG3 as the immunodominant region targeted by pathogenic antibodies in PV and indicate that autoreactivity relies on somatic mutations generated in the response to an antigen unrelated to DSG3.
Figures
References
-
- Ishii K, et al. Characterization of autoantibodies in pemphigus using antigen-specific enzyme-linked immunosorbent assays with baculovirus-expressed recombinant desmogleins. J Immunol. 1997;159(4):2010–2017. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
