

N-terminal protein processing: a comparative proteogenomic analysis
- PMID: 23001859
- PMCID: PMC3536895
- DOI: 10.1074/mcp.M112.019075
N-terminal protein processing: a comparative proteogenomic analysis
Abstract
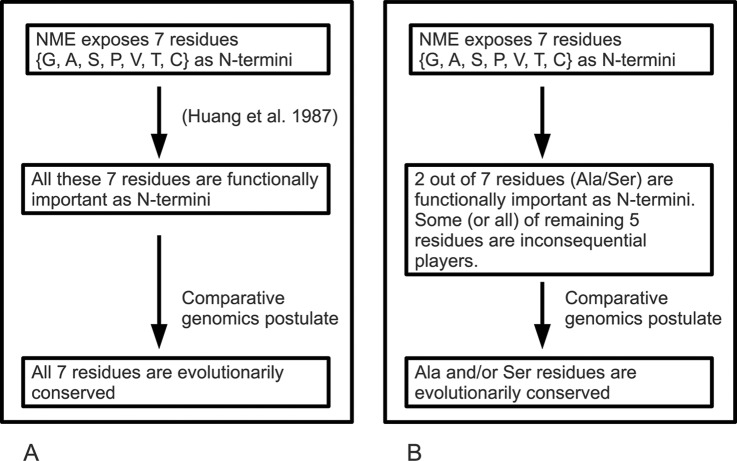
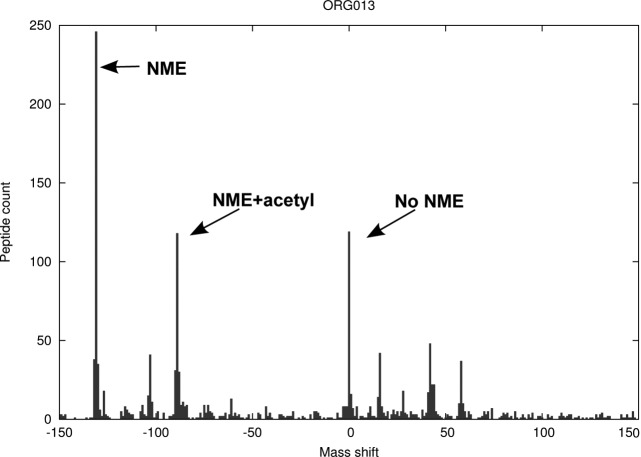
N-terminal methionine excision (NME) and N-terminal acetylation (NTA) are two of the most common protein post-translational modifications. NME is a universally conserved activity and a highly specific mechanism across all life forms. NTA is very common in eukaryotes but occurs rarely in prokaryotes. By analyzing data sets from yeast, mammals and bacteria (including 112 million spectra from 57 bacterial species), the largest comparative proteogenomics study to date, it is shown that previous assumptions/perceptions about the specificity and purposes of NME are not entirely correct. Although NME, through the universal enzymatic specificity of the methionine aminopeptidases, results in the removal of the initiator Met in proteins when the second residue is Gly, Ala, Ser, Cys, Thr, Pro, or Val, the comparative genomic analyses suggest that this specificity may vary modestly in some organisms. In addition, the functional role of NME may be primarily to expose Ala and Ser rather than all seven of these residues. Although any of this group provide "stabilizing" N termini in the N-end rule, and de facto leave the remaining 13 amino acid types that are classed as "destabilizing" (in higher eukaryotes) protected by the initiator Met, the conservation of NME-substrate proteins through evolution suggests that the other five are not crucially important for proteins with these residues in the second position. They are apparently merely inconsequential players (their function is not affected by NME) that become exposed because their side chains are smaller or comparable to those of Ala and Ser. The importance of exposing mainly two amino acids at the N terminus, i.e. Ala and Ser, is unclear but may be related to NTA or other post-translational modifications. In this regard, these analyses also reveal that NTA is more prevalent in some prokaryotes than previously appreciated.
Figures






Similar articles
-
N-terminal methionine excision of proteins creates tertiary destabilizing N-degrons of the Arg/N-end rule pathway.J Biol Chem. 2019 Mar 22;294(12):4464-4476. doi: 10.1074/jbc.RA118.006913. Epub 2019 Jan 23. J Biol Chem. 2019. PMID: 30674553 Free PMC article.
-
Characterization of N-terminal protein modifications in Pseudomonas aeruginosa PA14.J Proteomics. 2015 Jan 30;114:214-25. doi: 10.1016/j.jprot.2014.11.006. Epub 2014 Nov 21. J Proteomics. 2015. PMID: 25464366
-
Sequence determinants of cytosolic N-terminal protein processing.Eur J Biochem. 1986 Jan 2;154(1):193-6. doi: 10.1111/j.1432-1033.1986.tb09378.x. Eur J Biochem. 1986. PMID: 3080313
-
N-terminal processing: the methionine aminopeptidase and N alpha-acetyl transferase families.Trends Biochem Sci. 1998 Jul;23(7):263-7. doi: 10.1016/s0968-0004(98)01227-4. Trends Biochem Sci. 1998. PMID: 9697417 Review.
-
Protein alpha-N-acetylation studied by N-terminomics.FEBS J. 2011 Oct;278(20):3822-34. doi: 10.1111/j.1742-4658.2011.08230.x. Epub 2011 Aug 2. FEBS J. 2011. PMID: 21736701 Review.
Cited by
-
Quantitative N-Terminal Footprinting of Pathogenic Mycobacteria Reveals Differential Protein Acetylation.J Proteome Res. 2018 Sep 7;17(9):3246-3258. doi: 10.1021/acs.jproteome.8b00373. Epub 2018 Aug 16. J Proteome Res. 2018. PMID: 30080413 Free PMC article.
-
N-terminal methionine excision of proteins creates tertiary destabilizing N-degrons of the Arg/N-end rule pathway.J Biol Chem. 2019 Mar 22;294(12):4464-4476. doi: 10.1074/jbc.RA118.006913. Epub 2019 Jan 23. J Biol Chem. 2019. PMID: 30674553 Free PMC article.
-
Omics Assisted N-terminal Proteoform and Protein Expression Profiling On Methionine Aminopeptidase 1 (MetAP1) Deletion.Mol Cell Proteomics. 2018 Apr;17(4):694-708. doi: 10.1074/mcp.RA117.000360. Epub 2018 Jan 9. Mol Cell Proteomics. 2018. PMID: 29317475 Free PMC article.
-
Molecular and structural mechanisms of ZZ domain-mediated cargo selection by Nbr1.EMBO J. 2021 Aug 2;40(15):e107497. doi: 10.15252/embj.2020107497. Epub 2021 Jun 25. EMBO J. 2021. PMID: 34169534 Free PMC article.
-
N-terminomics reveals control of Arabidopsis seed storage proteins and proteases by the Arg/N-end rule pathway.New Phytol. 2018 May;218(3):1106-1126. doi: 10.1111/nph.14909. Epub 2017 Nov 23. New Phytol. 2018. PMID: 29168982 Free PMC article.
References
-
- Arfin S., Bradshaw R. (1988) Cotranslational processing and protein turnover in eukaryotic cells. Biochemistry 27, 7979–7984 - PubMed
-
- Frottin F., Martinez A., Peynot P., Mitra S., Holz R. C., Giglione C., Meinnel T. (2006) The Proteomics of N-terminal Methionine Cleavage. Mol. Cell. Proteomics 5, 2336–2349 - PubMed
-
- Meinnel T., Serero A., Giglione C. (2006) Impact of the N-terminal amino acid on targeted protein degradation. Biol. Chem. 387, 839–851 - PubMed
-
- Bradshaw R. A., Brickey W. W., Walker K. W. (1998) N-Terminal processing: the methionine aminopeptidase and N[alpha]-acetyl transferase families. Trends Biochem. Sci. 23, 263–267 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
