

Mitochondria in traumatic brain injury and mitochondrial-targeted multipotential therapeutic strategies
- PMID: 23003569
- PMCID: PMC3575772
- DOI: 10.1111/j.1476-5381.2012.02025.x
Mitochondria in traumatic brain injury and mitochondrial-targeted multipotential therapeutic strategies
Abstract
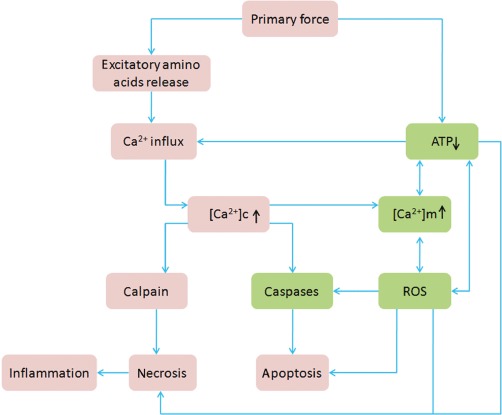
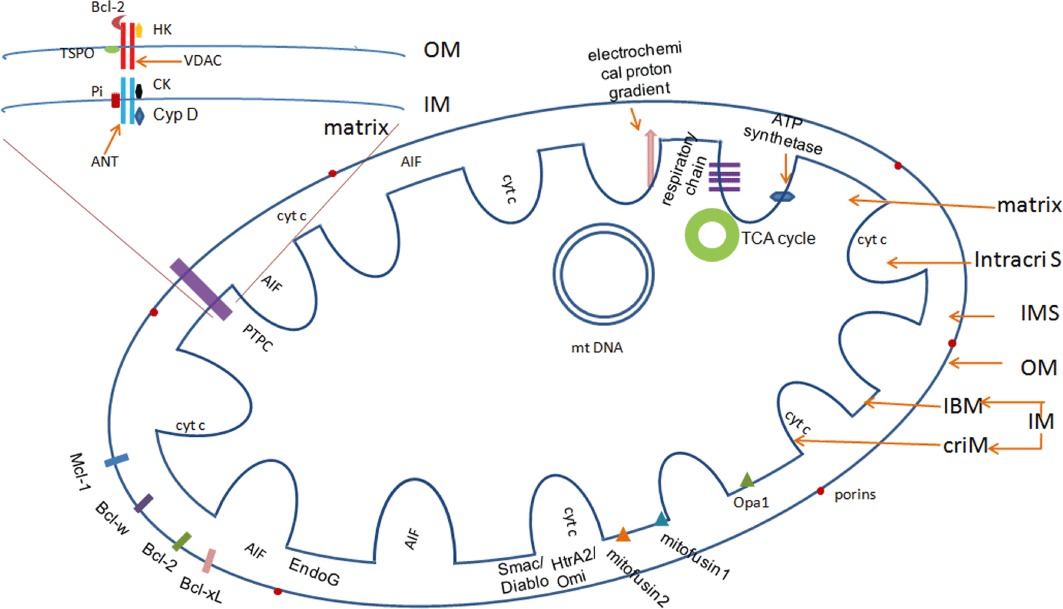
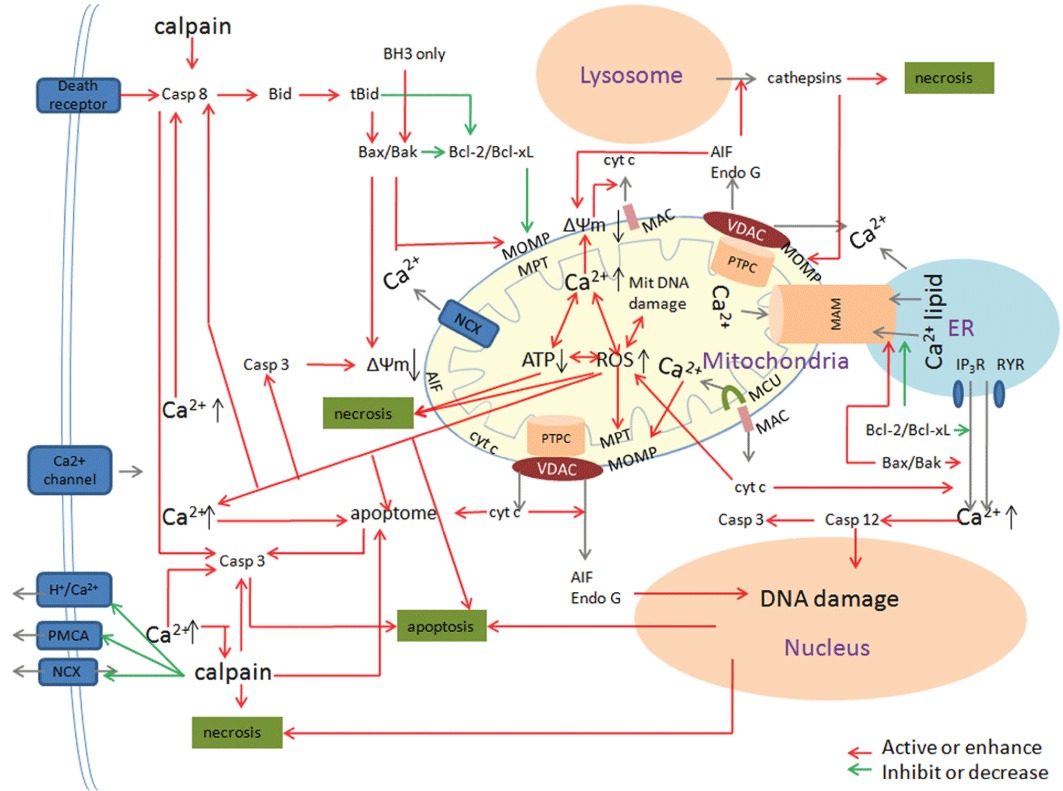
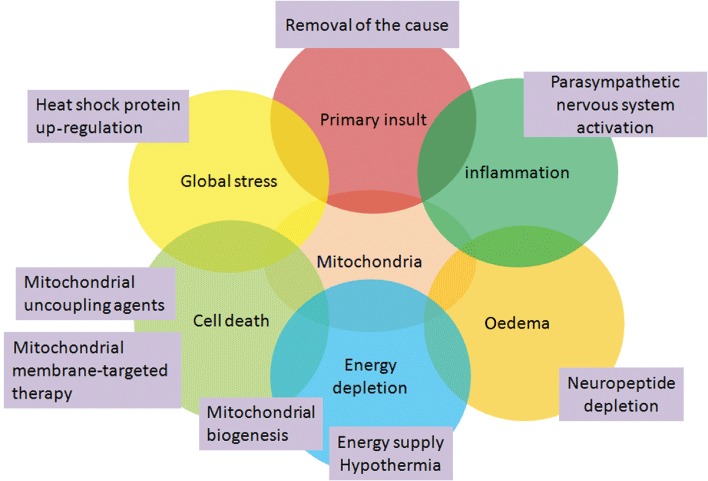
Traumatic brain injury (TBI) is a major health and socioeconomic problem throughout the world. It is a complicated pathological process that consists of primary insults and a secondary insult characterized by a set of biochemical cascades. The imbalance between a higher energy demand for repair of cell damage and decreased energy production led by mitochondrial dysfunction aggravates cell damage. At the cellular level, the main cause of the secondary deleterious cascades is cell damage that is centred in the mitochondria. Excitotoxicity, Ca(2+) overload, reactive oxygen species (ROS), Bcl-2 family, caspases and apoptosis inducing factor (AIF) are the main participants in mitochondria-centred cell damage following TBI. Some preclinical and clinical results of mitochondria-targeted therapy show promise. Mitochondria- targeted multipotential therapeutic strategies offer new hope for the successful treatment of TBI and other acute brain injuries.
© 2012 The Authors. British Journal of Pharmacology © 2012 The British Pharmacological Society.
Figures
References
-
- Albensi BC, Sullivan PG, Thompson MB, Scheff SW, Mattson MP. Cyclosporin ameliorates traumatic brain-injury-induced alterations of hippocampal synaptic plasticity. Exp Neurol. 2000;162:385–389. - PubMed
-
- Alessandri B, Rice AC, Levasseur J, DeFord M, Hamm RJ, Bullock MR. Cyclosporin A improves brain tissue oxygen consumption and learning/memory performance after lateral fluid percussion injury in rats. J Neurotrauma. 2002;19:829–841. - PubMed
-
- Anderson S, Bankier AT, Barrell BG, de Bruijn MH, Coulson AR, Drouin J, et al. Sequence and organization of the human mitochondrial genome. Nature. 1981;290:457–465. - PubMed
-
- Andreyev AY, Kushnareva YE, Starkov AA. Mitochondrial metabolism of reactive oxygen species. Biochemistry (Mosc) 2005;70:200–214. - PubMed
-
- Ankarcrona M, Dypbukt JM, Bonfoco E, Zhivotovsky B, Orrenius S, Lipton SA, et al. Glutamate-induced neuronal death: a succession of necrosis or apoptosis depending on mitochondrial function. Neuron. 1995;15:961–973. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
