

Cancer cell metabolism: one hallmark, many faces
- PMID: 23009760
- PMCID: PMC3491070
- DOI: 10.1158/2159-8290.CD-12-0345
Cancer cell metabolism: one hallmark, many faces
Abstract
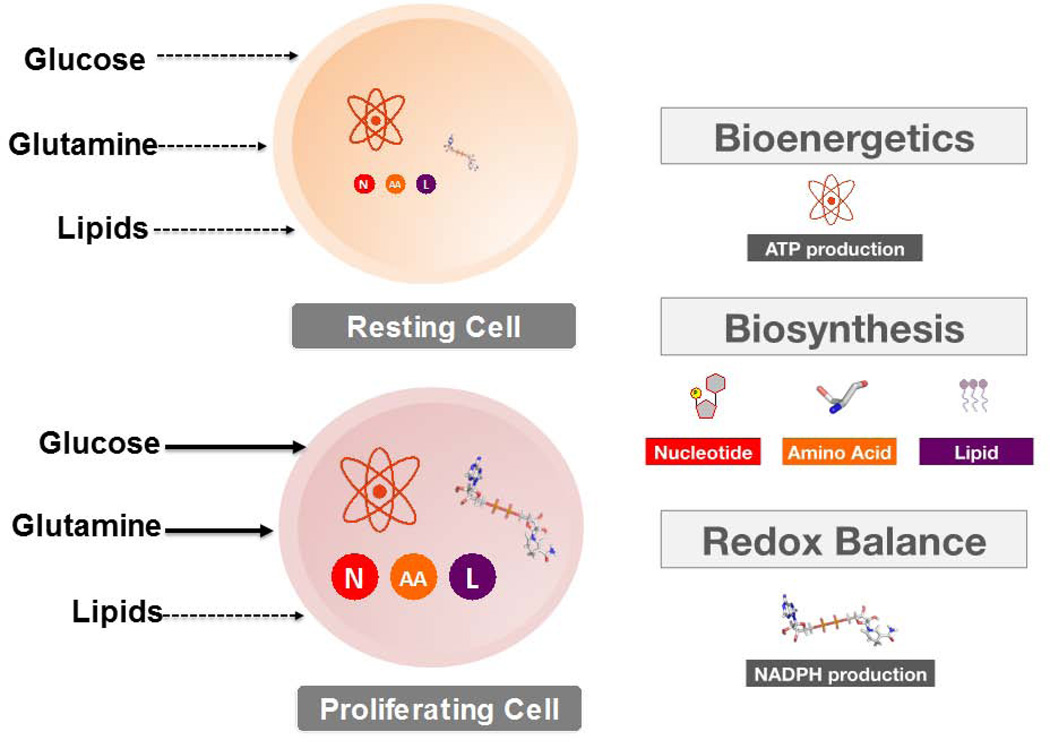
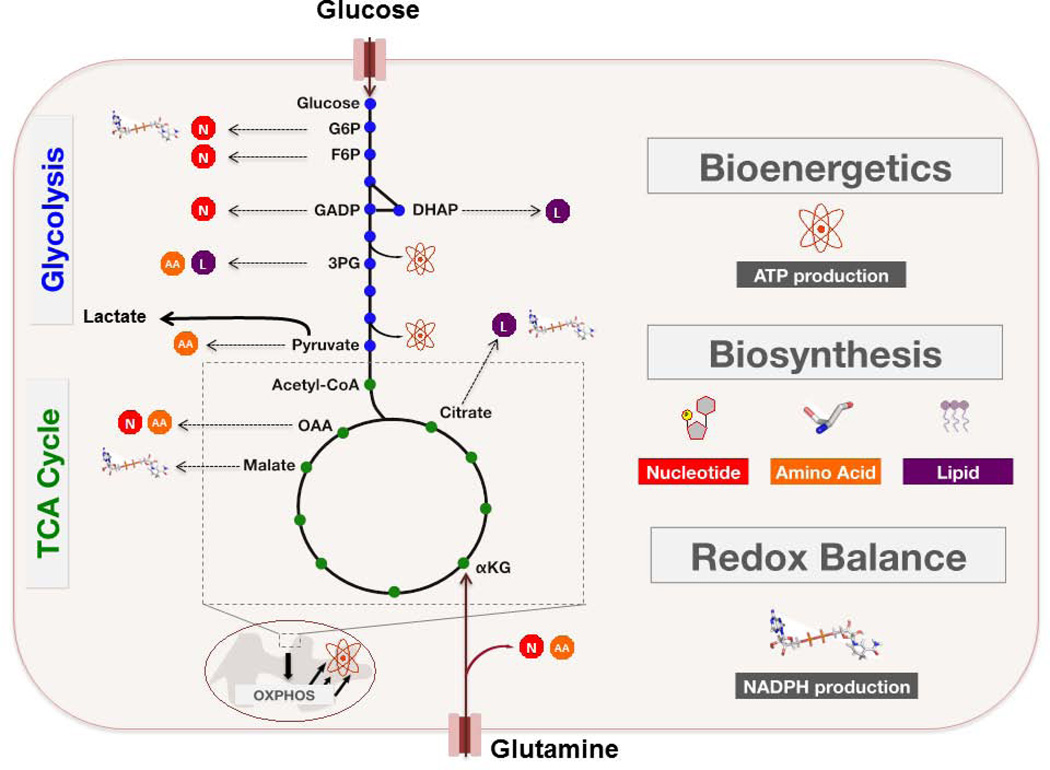
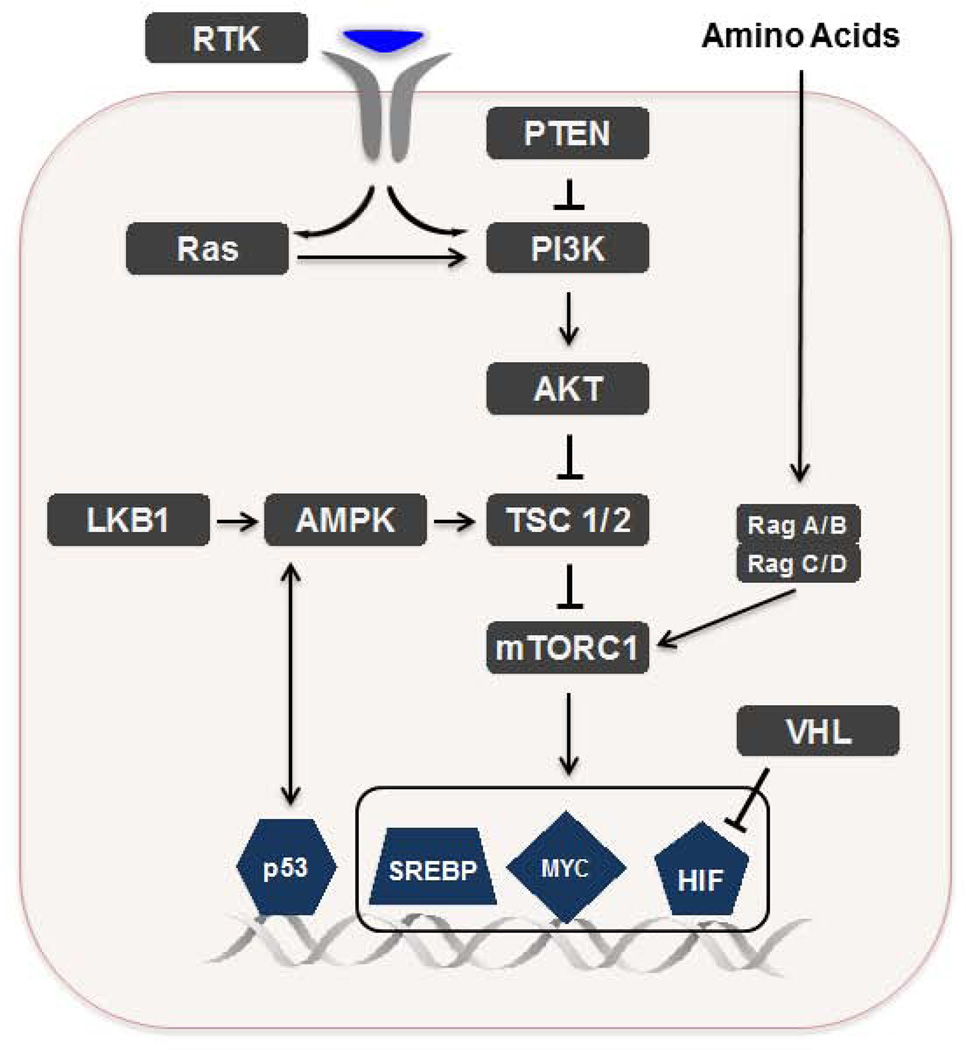
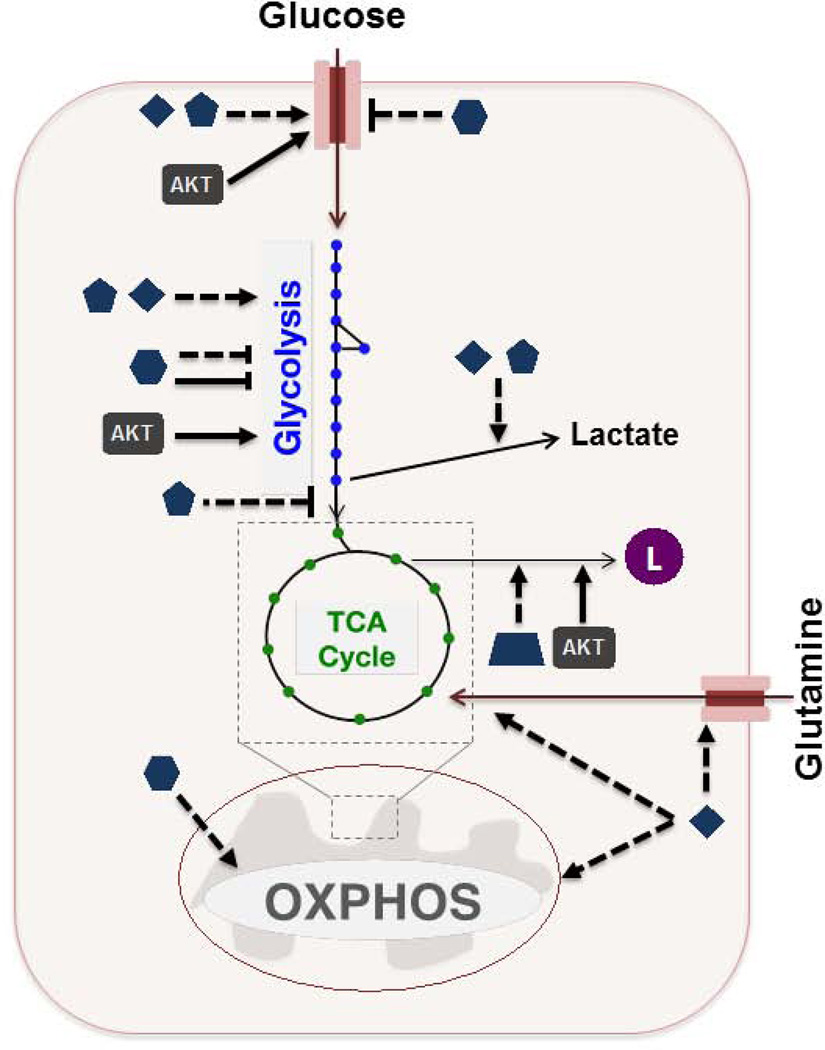
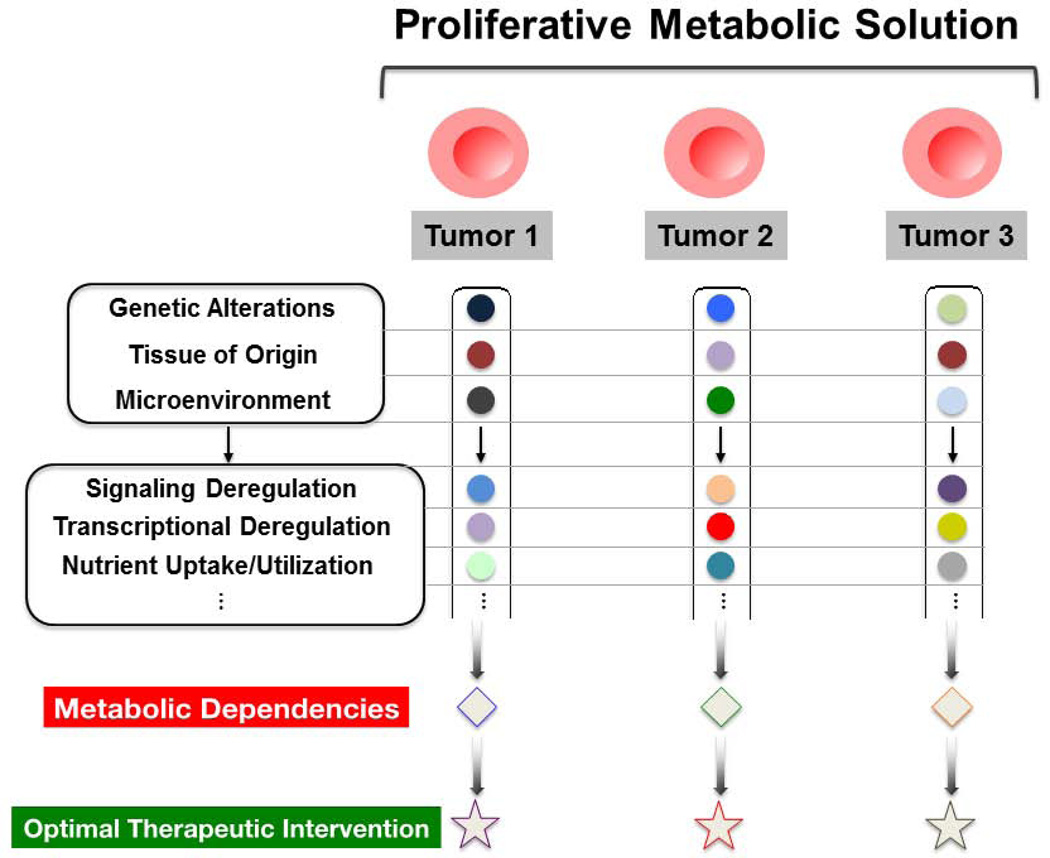
Cancer cells must rewire cellular metabolism to satisfy the demands of growth and proliferation. Although many of the metabolic alterations are largely similar to those in normal proliferating cells, they are aberrantly driven in cancer by a combination of genetic lesions and nongenetic factors such as the tumor microenvironment. However, a single model of altered tumor metabolism does not describe the sum of metabolic changes that can support cell growth. Instead, the diversity of such changes within the metabolic program of a cancer cell can dictate by what means proliferative rewiring is driven, and can also impart heterogeneity in the metabolic dependencies of the cell. A better understanding of this heterogeneity may enable the development and optimization of therapeutic strategies that target tumor metabolism.
Conflict of interest statement
The authors disclose no potential conflicts of interest.
Figures
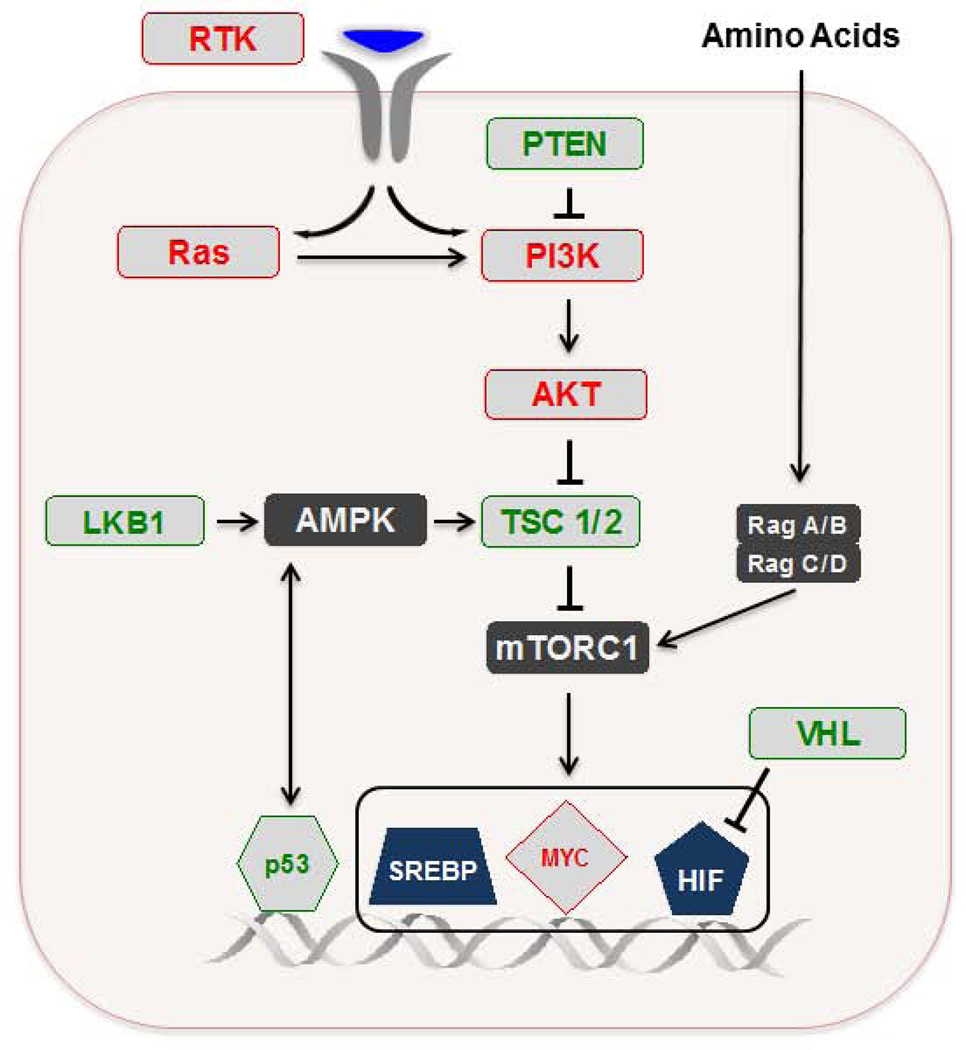
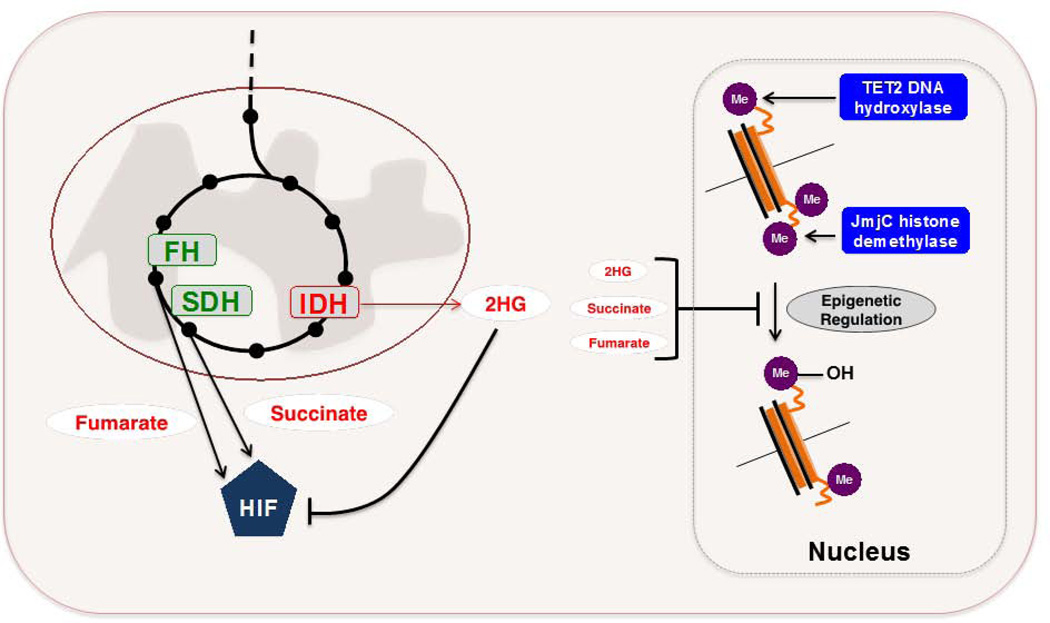
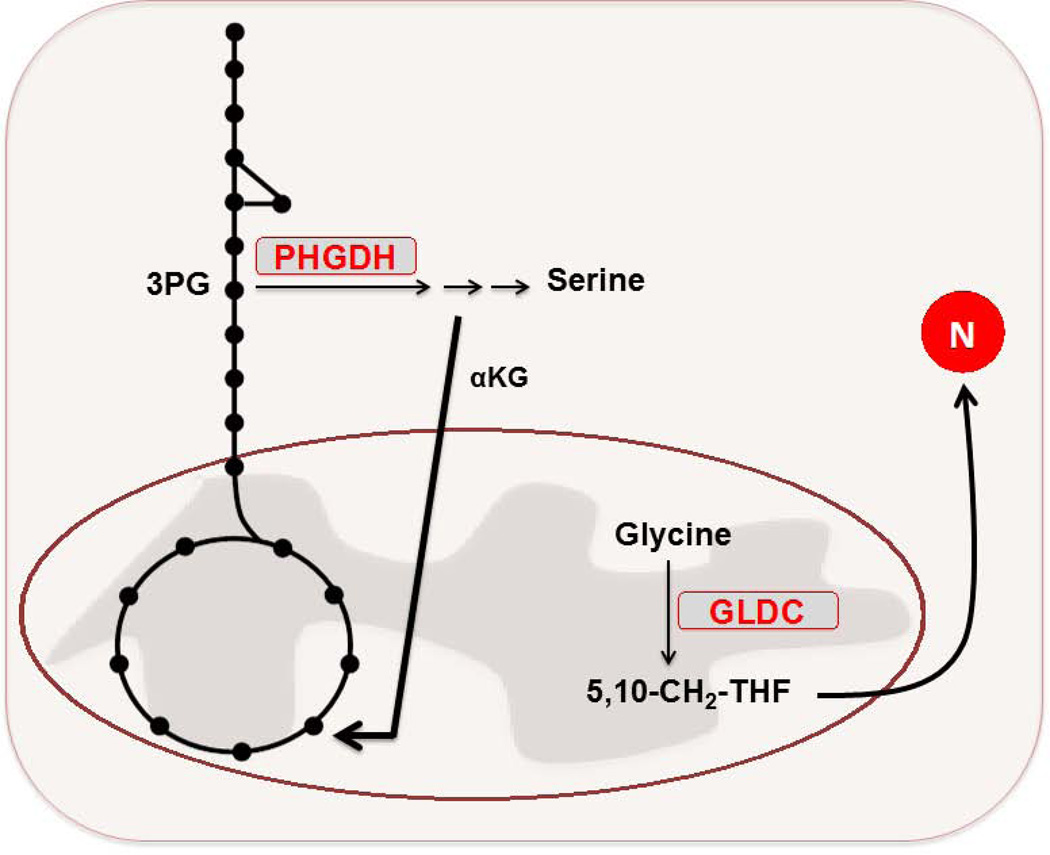
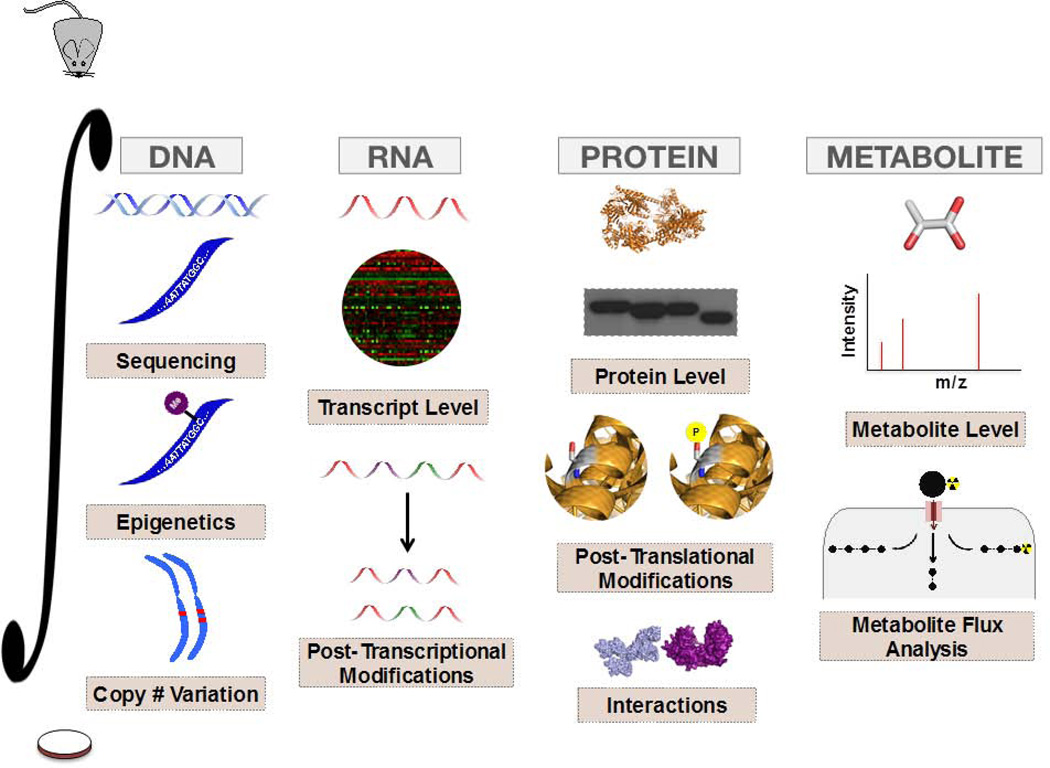
References
-
- Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin. 2012;62:10–29. - PubMed
-
- Kelloff GJ, Sigman CC. Cancer biomarkers: selecting the right drug for the right patient. Nat Rev Drug Discov. 2012;11:1–14. - PubMed
-
- Wong KM, Hudson TJ, McPherson JD. Unraveling the Genetics of Cancer: Genome Sequencing and Beyond. Annu Rev Genomics Hum Genet. 2011;12:407–430. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
