

Mitochondrial handling of excess Ca2+ is substrate-dependent with implications for reactive oxygen species generation
- PMID: 23010495
- PMCID: PMC3542420
- DOI: 10.1016/j.freeradbiomed.2012.09.020
Mitochondrial handling of excess Ca2+ is substrate-dependent with implications for reactive oxygen species generation
Abstract
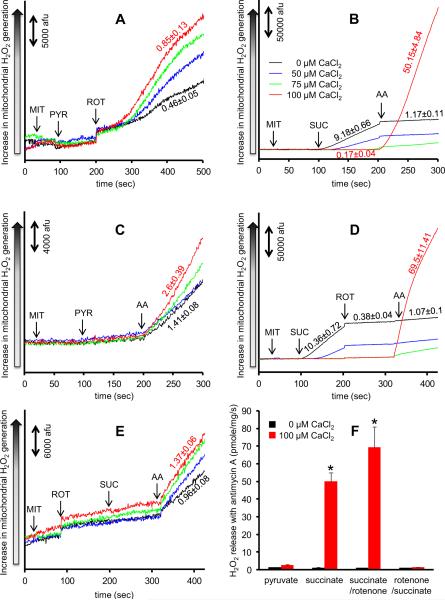
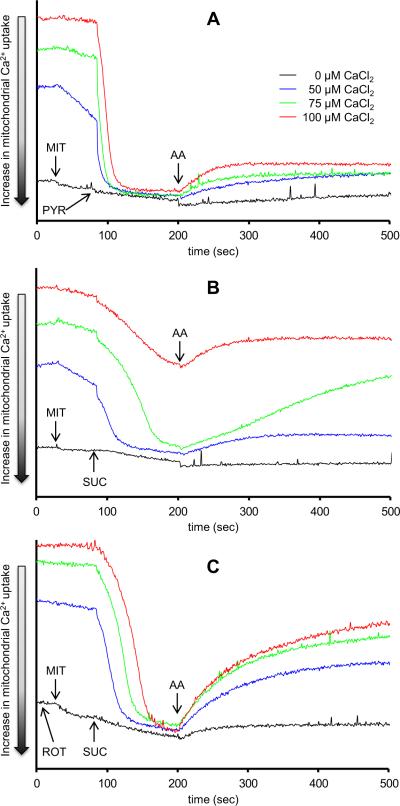
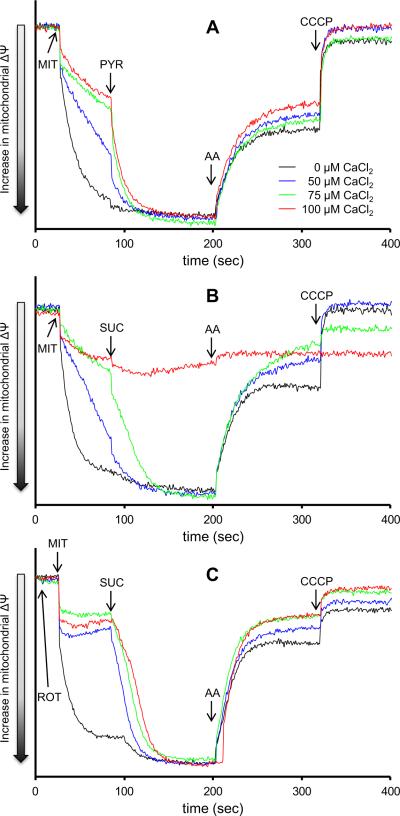
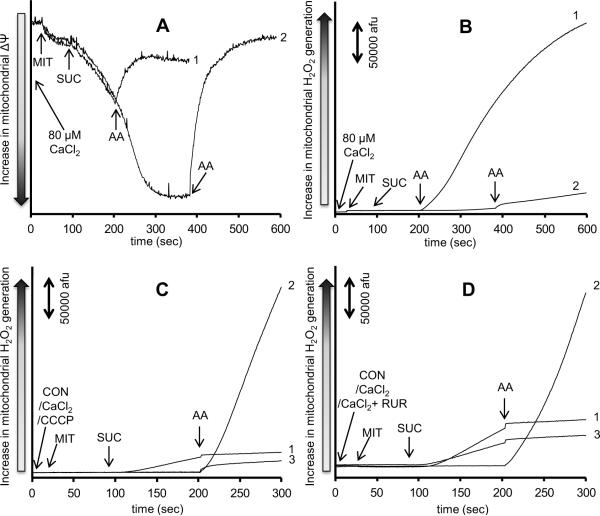
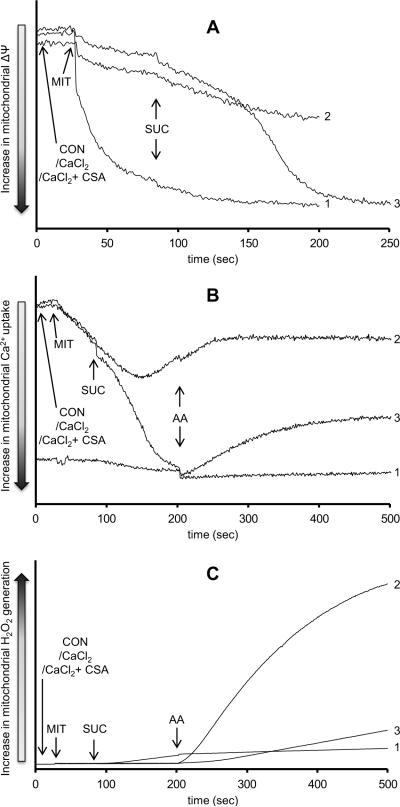
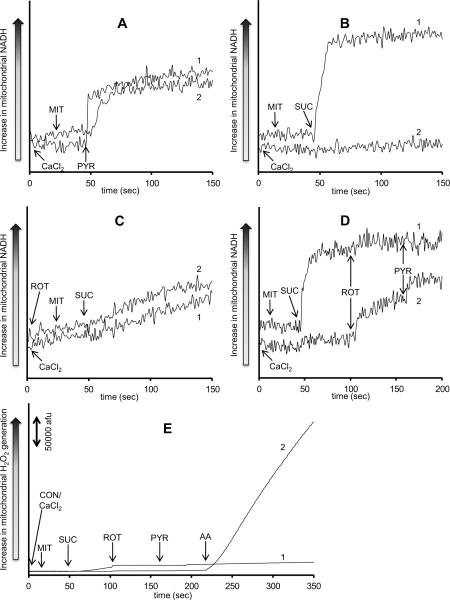
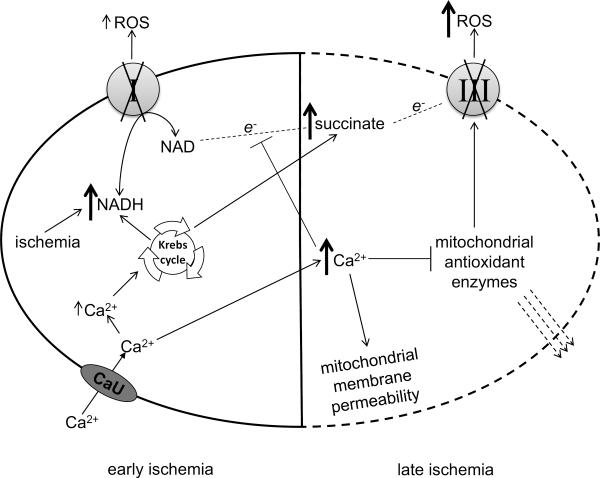
The mitochondrial electron transport chain is the major source of reactive oxygen species (ROS) during cardiac ischemia. Several mechanisms modulate ROS production; one is mitochondrial Ca(2+) uptake. Here we sought to elucidate the effects of extramitochondrial Ca(2+) (e[Ca(2+)]) on ROS production (measured as H(2)O(2) release) from complexes I and III. Mitochondria isolated from guinea pig hearts were preincubated with increasing concentrations of CaCl(2) and then energized with the complex I substrate Na(+) pyruvate or the complex II substrate Na(+) succinate. Mitochondrial H(2)O(2) release rates were assessed after giving either rotenone or antimycin A to inhibit complex I or III, respectively. After pyruvate, mitochondria maintained a fully polarized membrane potential (ΔΨ; assessed using rhodamine 123) and were able to generate NADH (assessed using autofluorescence) even with excess e[Ca(2+)] (assessed using CaGreen-5N), whereas they remained partially depolarized and did not generate NADH after succinate. This partial ΔΨ depolarization with succinate was accompanied by a large release in H(2)O(2) (assessed using Amplex red/horseradish peroxidase) with later addition of antimycin A. In the presence of excess e[Ca(2+)], adding cyclosporin A to inhibit mitochondrial permeability transition pore opening restored ΔΨ and significantly decreased antimycin A-induced H(2)O(2) release. Succinate accumulates during ischemia to become the major substrate utilized by cardiac mitochondria. The inability of mitochondria to maintain a fully polarized ΔΨ under excess e[Ca(2+)] when succinate, but not pyruvate, is the substrate may indicate a permeabilization of the mitochondrial membrane, which enhances H(2)O(2) emission from complex III during ischemia.
Copyright © 2012 Elsevier Inc. All rights reserved.
Figures
References
-
- Tsutsui H, Kinugawa S, Matsushima S. Oxidative stress and mitochondrial DNA damage in heart failure. Circ J. 2008;72(Suppl A):A31–37. - PubMed
-
- Stone D, Darley-Usmar V, Smith DR, O'Leary V. Hypoxia-reoxygenation induced increase in cellular Ca2+ in myocytes and perfused hearts: the role of mitochondria. J Mol Cell Cardiol. 1989;21:963–973. - PubMed
-
- Halestrap AP, Clarke SJ, Javadov SA. Mitochondrial permeability transition pore opening during myocardial reperfusion--a target for cardioprotection. Cardiovasc Res. 2004;61:372–385. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
