

Mutations in the INF2 gene account for a significant proportion of familial but not sporadic focal and segmental glomerulosclerosis
- PMID: 23014460
- PMCID: PMC3647680
- DOI: 10.1038/ki.2012.349
Mutations in the INF2 gene account for a significant proportion of familial but not sporadic focal and segmental glomerulosclerosis
Abstract
Mutations in the inverted formin 2 gene (INF2) have recently been identified as the most common cause of autosomal dominant focal and segmental glomerulosclerosis (FSGS). To quantify the contribution of various genes contributing to FSGS, we sequenced INF2 where all mutations have previously been described (exons 2 to 5) in a total of 215 probands and 281 sporadic individuals with FSGS, along with other known genes accounting for autosomal dominant FSGS (ACTN4, TRPC6, and CD2AP) in 213 probands. Variants were classified as disease-causing if they altered the amino acid sequence and if they were not found in control samples and in families segregated with disease. Mutations in INF2 were found in a total of 20 of the 215 families (including those previously reported) in our cohort of autosomal dominant familial nephrotic syndrome or FSGS, thereby explaining disease in 9%. INF2 mutations were found in 2 of 281 individuals with sporadic FSGS. In contrast, ACTN4- and TRPC6-related diseases accounted for 3 and 2% of our familial cohort, respectively. INF2-related disease showed variable penetrance, with onset of disease ranging widely from childhood to adulthood, and commonly leading to end-stage renal disease in the third and fourth decade of life. Thus, mutations in INF2 are a more common, although still a minor, monogenic cause of familial FSGS when compared with other known autosomal dominant genes associated with FSGS.
Figures

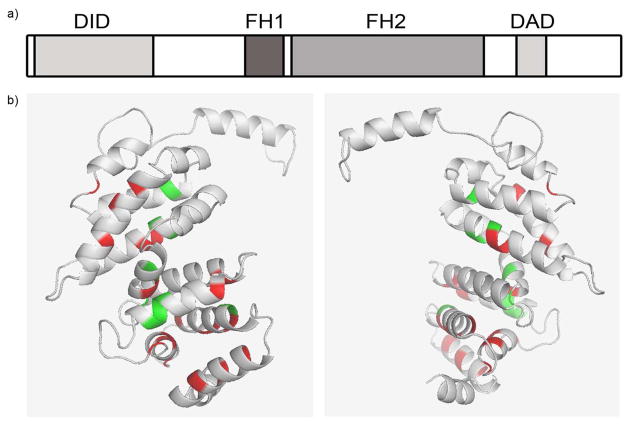
References
-
- Simon P, Ramee MP, Boulahrouz R, et al. Epidemiologic data of primary glomerular diseases in western France. Kidney international. 2004;66:905–908. - PubMed
-
- Briganti EM, Dowling J, Finlay M, et al. The incidence of biopsy-proven glomerulonephritis in Australia. Nephrology, dialysis, transplantation: official publication of the European Dialysis and Transplant Association - European Renal Association. 2001;16:1364–1367. - PubMed
-
- Haas M, Spargo BH, Coventry S. Increasing incidence of focal-segmental glomerulosclerosis among adult nephropathies: a 20-year renal biopsy study. American journal of kidney diseases: the official journal of the National Kidney Foundation. 1995;26:740–750. - PubMed
-
- Kitiyakara C, Eggers P, Kopp JB. Twenty-one-year trend in ESRD due to focal segmental glomerulosclerosis in the United States. American journal of kidney diseases: the official journal of the National Kidney Foundation. 2004;44:815–825. - PubMed
-
- Kaplan JM, Kim SH, North KN, et al. Mutations in ACTN4, encoding alpha-actinin-4, cause familial focal segmental glomerulosclerosis. Nature genetics. 2000;24:251–256. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
- R01 DK054931/DK/NIDDK NIH HHS/United States
- RC2 HL102926/HL/NHLBI NIH HHS/United States
- CAPMC/ CIHR/Canada
- R56 DK054931/DK/NIDDK NIH HHS/United States
- HL-102924/HL/NHLBI NIH HHS/United States
- RC2 HL102924/HL/NHLBI NIH HHS/United States
- HL-102926/HL/NHLBI NIH HHS/United States
- HL-102925/HL/NHLBI NIH HHS/United States
- HL-102923/HL/NHLBI NIH HHS/United States
- RC2 HL102923/HL/NHLBI NIH HHS/United States
- UC2 HL102926/HL/NHLBI NIH HHS/United States
- UC2 HL103010/HL/NHLBI NIH HHS/United States
- HL-103010/HL/NHLBI NIH HHS/United States
- K12 HD052896/HD/NICHD NIH HHS/United States
- DK54931/DK/NIDDK NIH HHS/United States
- UC2 HL102923/HL/NHLBI NIH HHS/United States
- UC2 HL102924/HL/NHLBI NIH HHS/United States
- RC2 HL103010/HL/NHLBI NIH HHS/United States
- 5K12HDO52896/PHS HHS/United States
- RC2 HL102925/HL/NHLBI NIH HHS/United States
- UC2 HL102925/HL/NHLBI NIH HHS/United States
- R01 DK088826/DK/NIDDK NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
