

Null mutations at the p66 and bradykinin 2 receptor loci induce divergent phenotypes in the diabetic kidney
- PMID: 23019230
- PMCID: PMC3532473
- DOI: 10.1152/ajprenal.00246.2012
Null mutations at the p66 and bradykinin 2 receptor loci induce divergent phenotypes in the diabetic kidney
Abstract
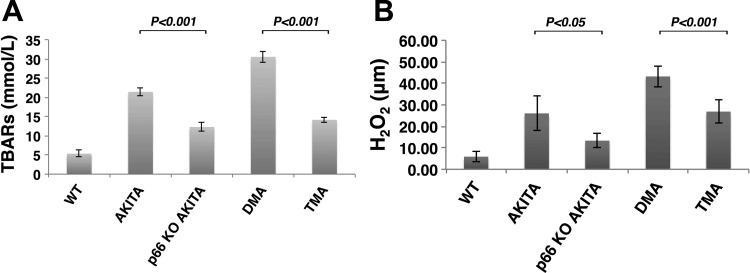
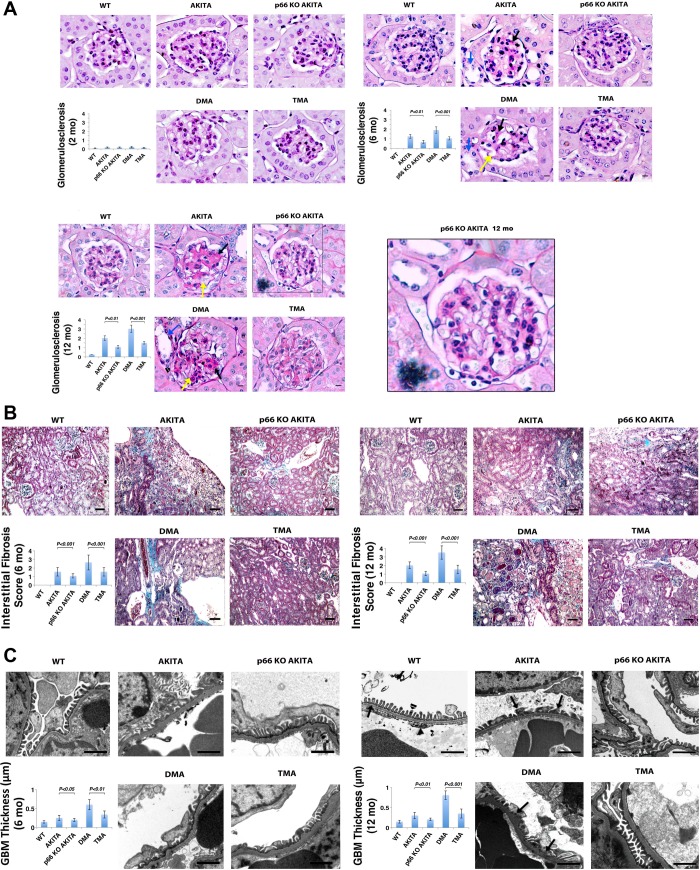
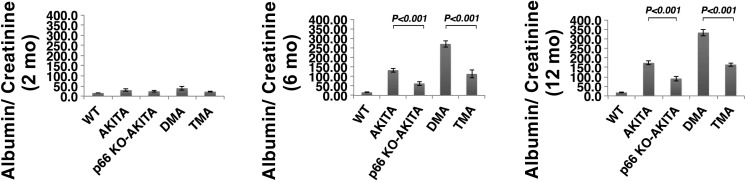
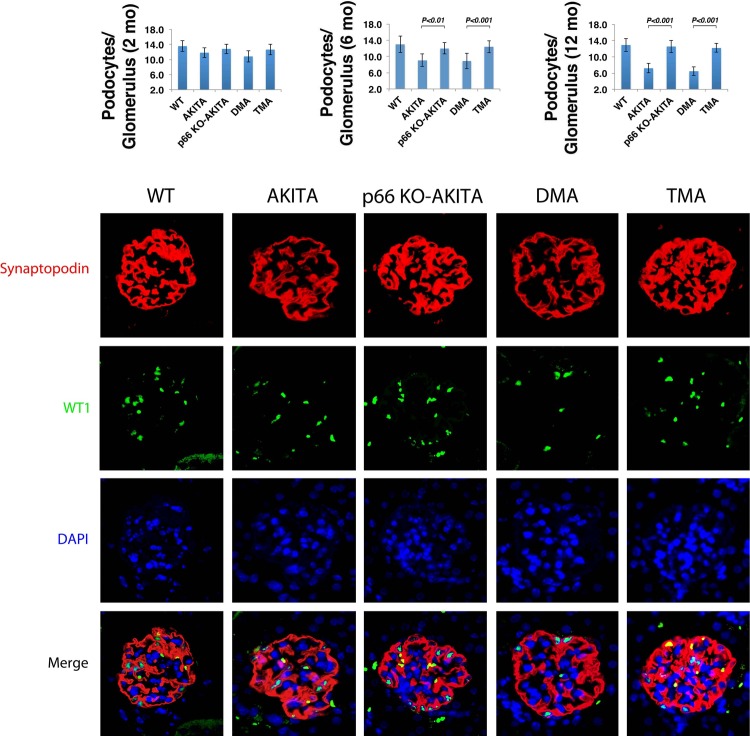
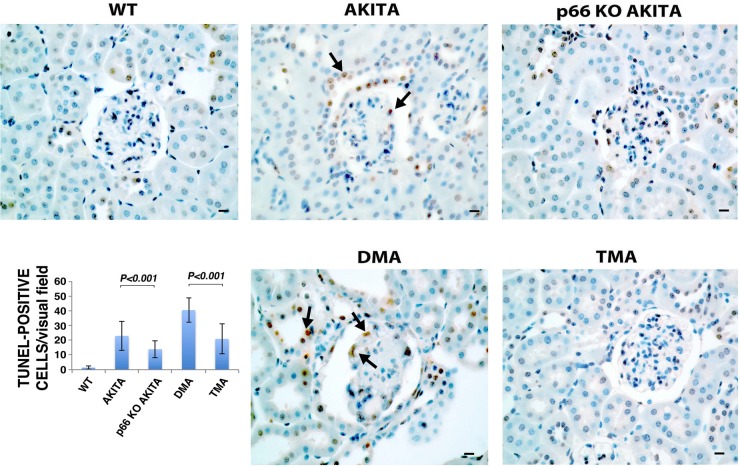
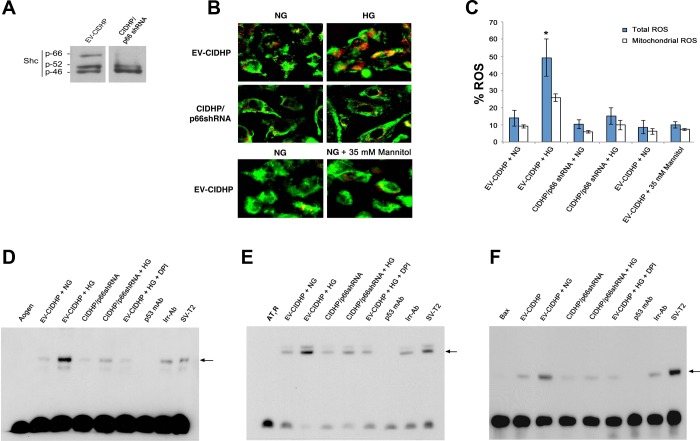
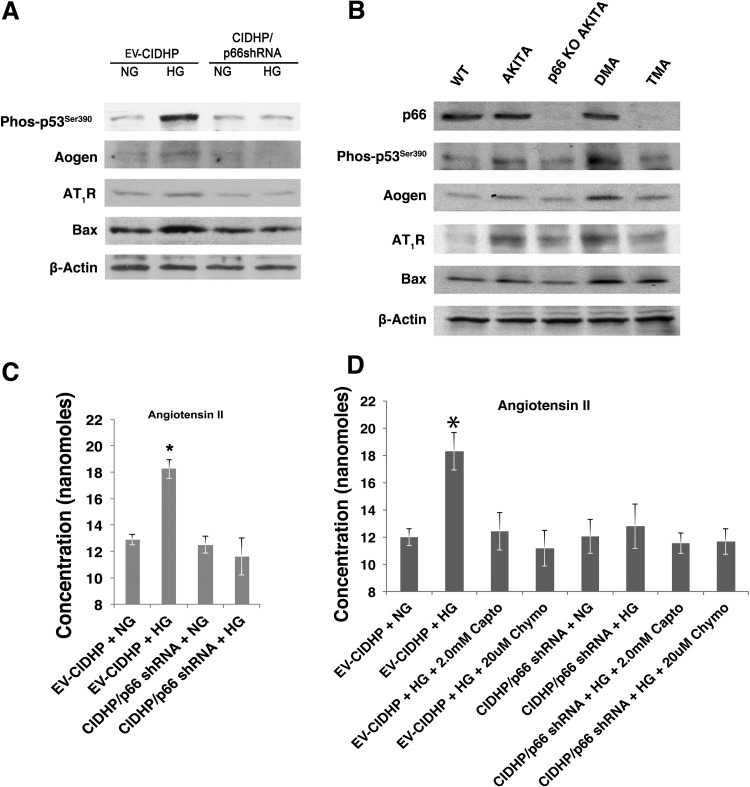
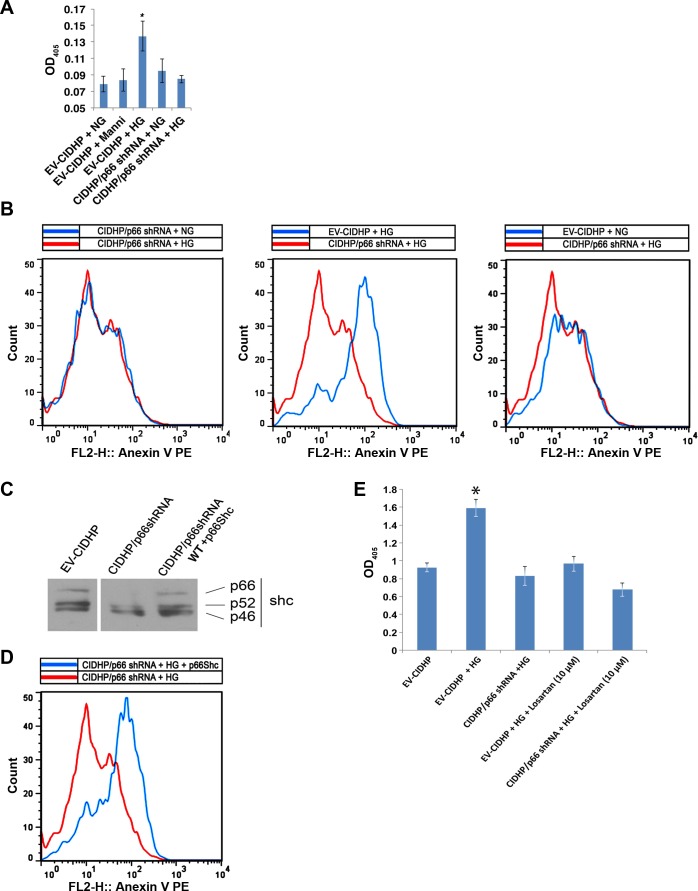
Candidate genes have been identified that confer increased risk for diabetic glomerulosclerosis (DG). Mice heterozygous for the Akita (Ins2(+/C96Y)) diabetogenic mutation with a second mutation introduced at the bradykinin 2 receptor (B2R(-/-)) locus express a disease phenotype that approximates human DG. Src homology 2 domain transforming protein 1 (p66) controls mitochondrial metabolism and cellular responses to oxidative stress, aging, and apoptosis. We generated p66-null Akita mice to test whether inactivating mutations at the p66 locus will rescue kidneys of Akita mice from disease-causing mutations at the Ins2 and B2R loci. Here we show null mutations at the p66 and B2R loci interact with the Akita (Ins2(+/C96Y)) mutation, independently and in combination, inducing divergent phenotypes in the kidney. The B2R(-/-) mutation induces detrimental phenotypes, as judged by increased systemic and renal levels of oxidative stress, histology, and urine albumin excretion, whereas the p66-null mutation confers a powerful protection phenotype. To elucidate the mechanism(s) of the protection phenotype, we turned to our in vitro system. Experiments with cultured podocytes revealed previously unrecognized cross talk between p66 and the redox-sensitive transcription factor p53 that controls hyperglycemia-induced ROS metabolism, transcription of p53 target genes (angiotensinogen, angiotensin II type-1 receptor, and bax), angiotensin II generation, and apoptosis. RNA-interference targeting p66 inhibits all of the above. Finally, protein levels of p53 target genes were upregulated in kidneys of Akita mice but unchanged in p66-null Akita mice. Taken together, p66 is a potential molecular target for therapeutic intervention in DG.
Figures
References
-
- Brenner BM, Cooper ME, de Zeeuw D, Keane WF, Mitch WE, Parving HH, Remuzzi G, Snapinn SM, Zhang Z, Shahinfar S. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med 345: 861–869, 2001 - PubMed
-
- Camici GG, Schiavoni M, Francia P, Bachschmid M, Martin-Padura I, Hersberger M, Tanner FC, Pelicci P, Volpe M, Anversa P, Luscher TF, Cosentino F. Genetic deletion of p66(Shc) adaptor protein prevents hyperglycemia-induced endothelial dysfunction and oxidative stress. Proc Natl Acad Sci USA 104: 5217–5222, 2007 - PMC - PubMed
-
- Chintapalli J, Yang S, Opawumi D, Goyal SR, Shamsuddin N, Malhotra A, Reiss K, Meggs LG. Inhibition of wild-type p66ShcA in mesangial cells prevents glycooxidant-dependent FOXO3a regulation and promotes the survival phenotype. Am J Physiol Renal Physiol 292: F523–F530, 2007 - PubMed
-
- Durvasula RV, Shankland SJ. Activation of a local renin angiotensin system in podocytes by glucose. Am J Physiol Renal Physiol 294: F830–F839, 2008 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous