

Ciliary neurotrophic factor activates NF-κB to enhance mitochondrial bioenergetics and prevent neuropathy in sensory neurons of streptozotocin-induced diabetic rodents
- PMID: 23022047
- PMCID: PMC3521091
- DOI: 10.1016/j.neuropharm.2012.09.015
Ciliary neurotrophic factor activates NF-κB to enhance mitochondrial bioenergetics and prevent neuropathy in sensory neurons of streptozotocin-induced diabetic rodents
Abstract
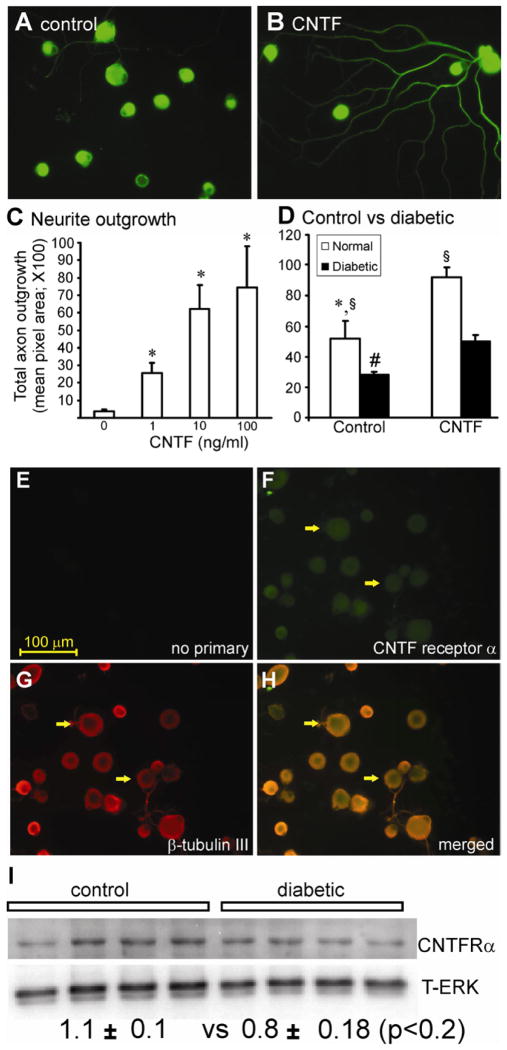
Diabetes causes mitochondrial dysfunction in sensory neurons that may contribute to peripheral neuropathy. Ciliary neurotrophic factor (CNTF) promotes sensory neuron survival and axon regeneration and prevents axonal dwindling, nerve conduction deficits and thermal hypoalgesia in diabetic rats. In this study, we tested the hypothesis that CNTF protects sensory neuron function during diabetes through normalization of impaired mitochondrial bioenergetics. In addition, we investigated whether the NF-κB signal transduction pathway was mobilized by CNTF. Neurite outgrowth of sensory neurons derived from streptozotocin (STZ)-induced diabetic rats was reduced compared to neurons from control rats and exposure to CNTF for 24 h enhanced neurite outgrowth. CNTF also activated NF-κB, as assessed by Western blotting for the NF-κB p50 subunit and reporter assays for NF-κB promoter activity. Conversely, blockade of NF-κB signaling using SN50 peptide inhibited CNTF-mediated neurite outgrowth. Studies in mice with STZ-induced diabetes demonstrated that systemic therapy with CNTF prevented functional indices of peripheral neuropathy along with deficiencies in dorsal root ganglion (DRG) NF-κB p50 expression and DNA binding activity. DRG neurons derived from STZ-diabetic mice also exhibited deficiencies in maximal oxygen consumption rate and associated spare respiratory capacity that were corrected by exposure to CNTF for 24 h in an NF-κB-dependent manner. We propose that the ability of CNTF to enhance axon regeneration and protect peripheral nerve from structural and functional indices of diabetic peripheral neuropathy is associated with targeting of mitochondrial function, in part via NF-κB activation, and improvement of cellular bioenergetics.
Copyright © 2012 Elsevier Ltd. All rights reserved.
Figures




References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
