

Site identification in high-throughput RNA-protein interaction data
- PMID: 23024010
- PMCID: PMC3509493
- DOI: 10.1093/bioinformatics/bts569
Site identification in high-throughput RNA-protein interaction data
Abstract
Motivation: Post-transcriptional and co-transcriptional regulation is a crucial link between genotype and phenotype. The central players are the RNA-binding proteins, and experimental technologies [such as cross-linking with immunoprecipitation- (CLIP-) and RIP-seq] for probing their activities have advanced rapidly over the course of the past decade. Statistically robust, flexible computational methods for binding site identification from high-throughput immunoprecipitation assays are largely lacking however.
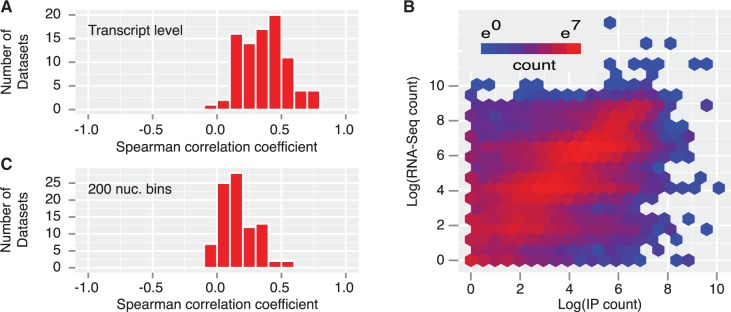
Results: We introduce a method for site identification which provides four key advantages over previous methods: (i) it can be applied on all variations of CLIP and RIP-seq technologies, (ii) it accurately models the underlying read-count distributions, (iii) it allows external covariates, such as transcript abundance (which we demonstrate is highly correlated with read count) to inform the site identification process and (iv) it allows for direct comparison of site usage across cell types or conditions.
Availability and implementation: We have implemented our method in a software tool called Piranha. Source code and binaries, licensed under the GNU General Public License (version 3) are freely available for download from http://smithlab.usc.edu.
Contact: andrewds@usc.edu
Supplementary information: Supplementary data available at Bioinformatics online.
Figures


References
-
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. B Methodological. 1995;57:289–300.
-
- Cameron AC, Trivedi PK. Regression Analysis of Count Data. Cambridge MA, UK: Cambridge University Press; 2008.
-
- Chénard CA, Richard S. New implications for the QUAKING RNA binding protein in human disease. J. Neurosci. Res. 2008;86:233–242. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
