

Somatic Populations of PGT135-137 HIV-1-Neutralizing Antibodies Identified by 454 Pyrosequencing and Bioinformatics
- PMID: 23024643
- PMCID: PMC3441199
- DOI: 10.3389/fmicb.2012.00315
Somatic Populations of PGT135-137 HIV-1-Neutralizing Antibodies Identified by 454 Pyrosequencing and Bioinformatics
Abstract
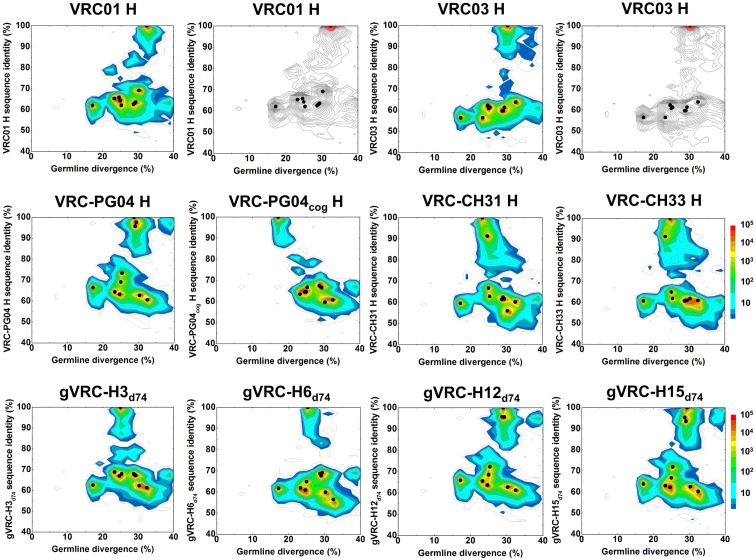
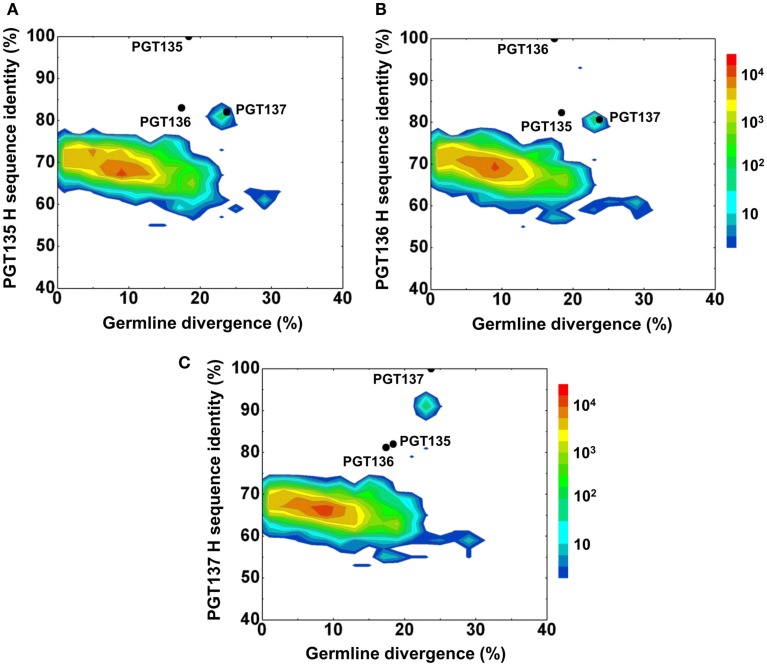
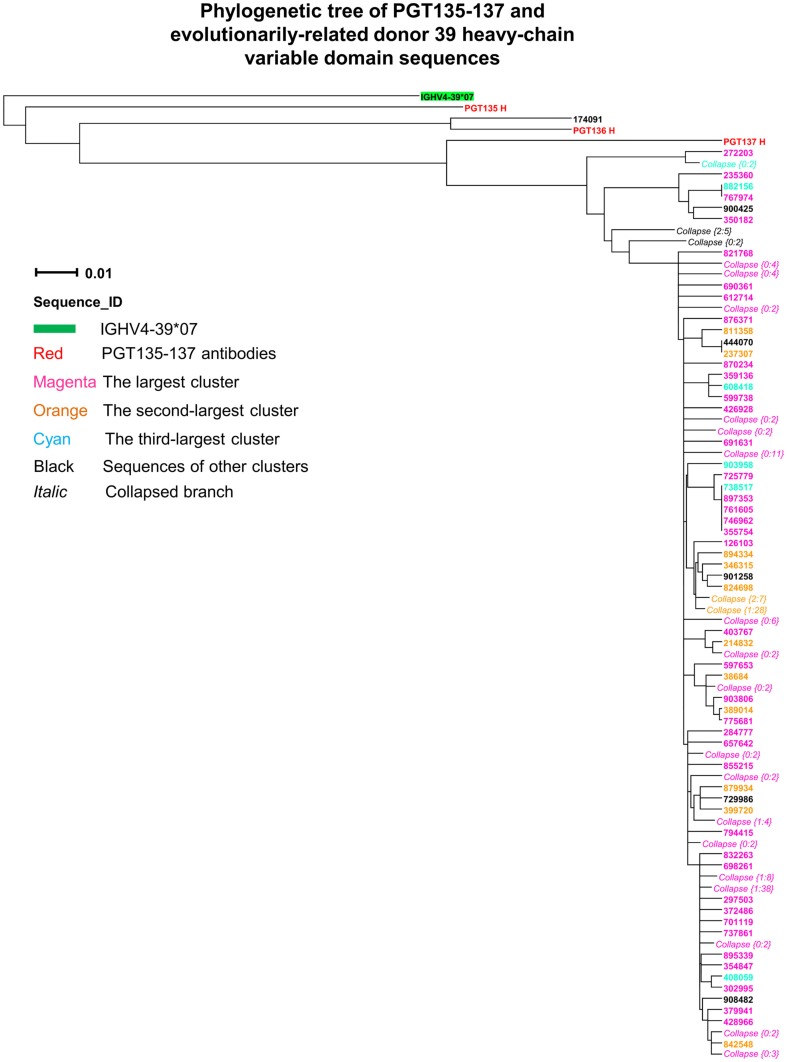
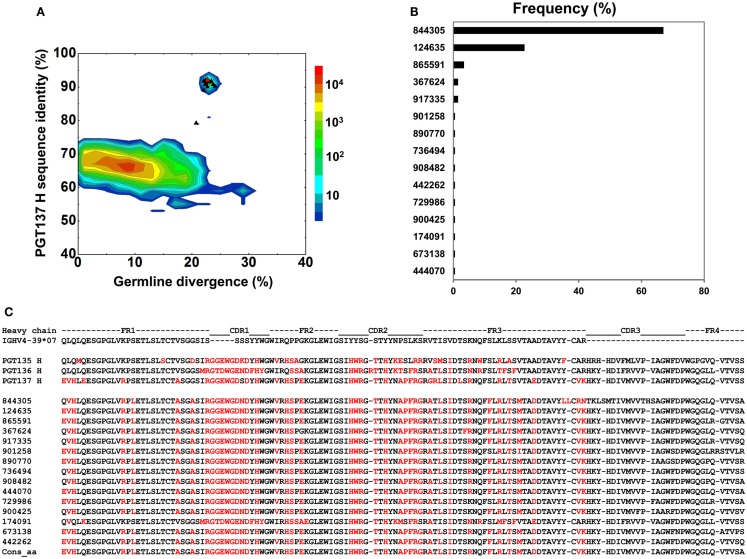
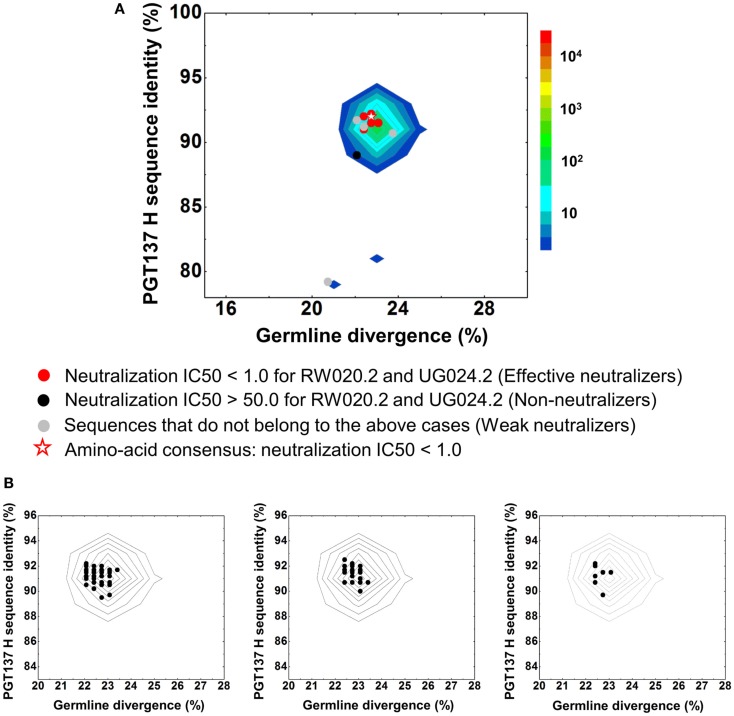
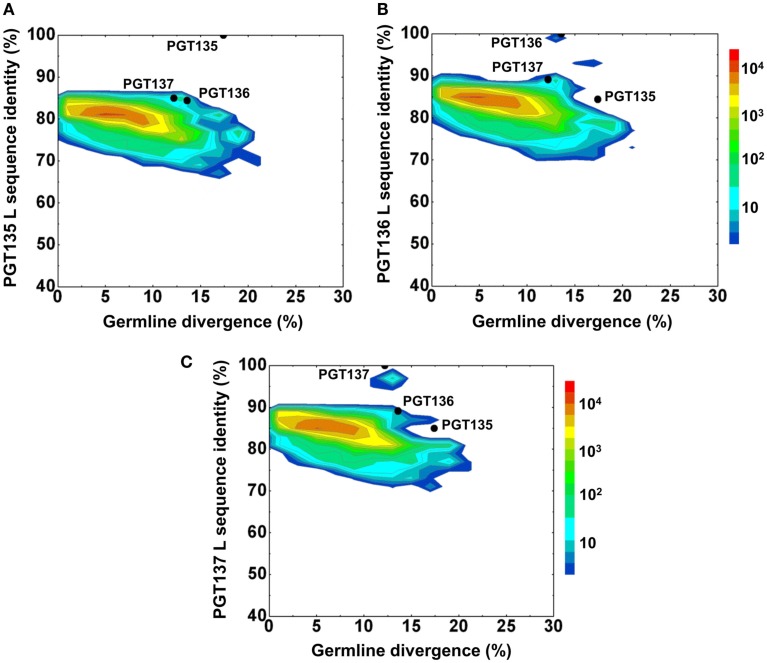
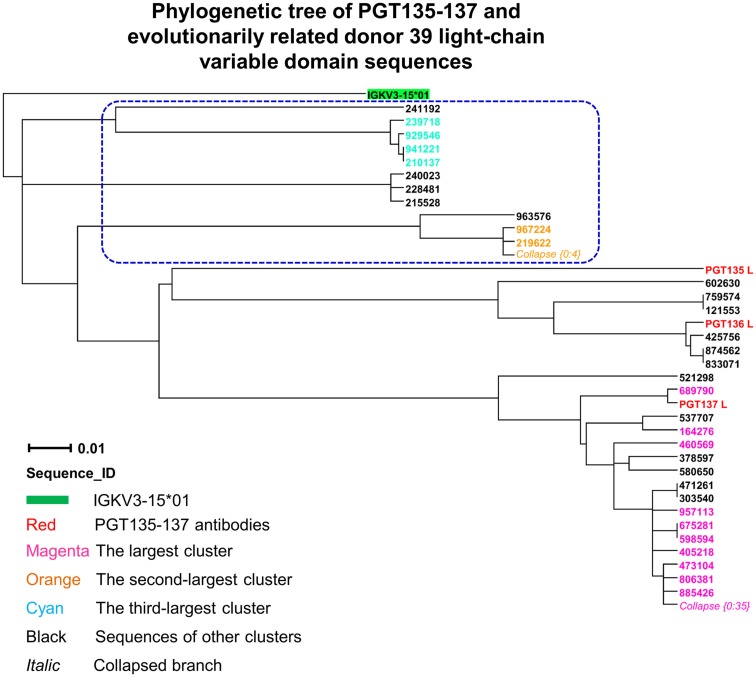
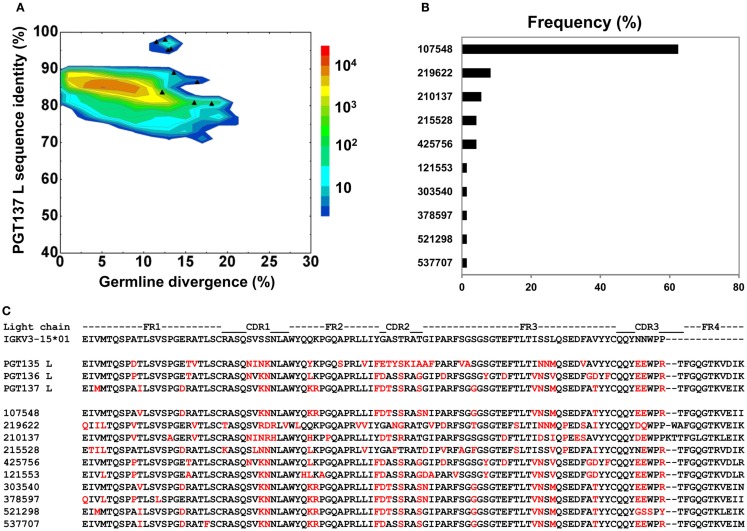
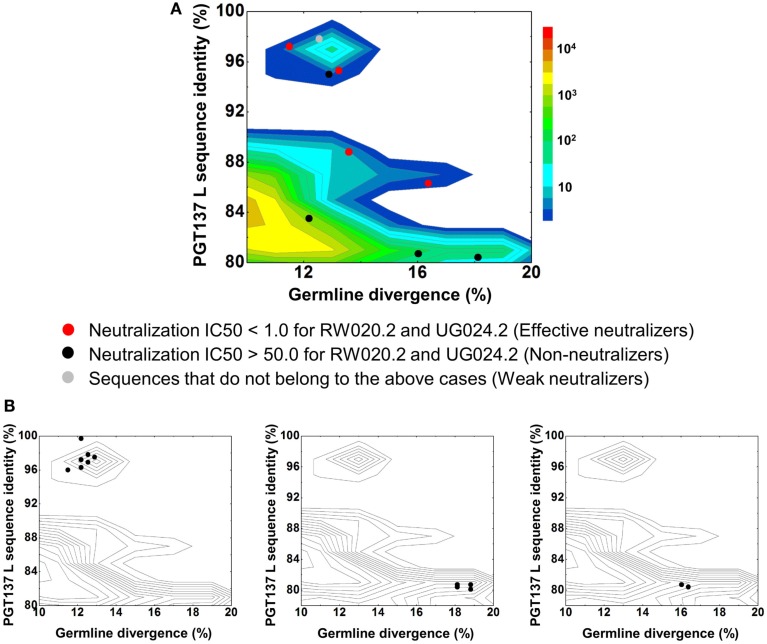
Select HIV-1-infected individuals develop sera capable of neutralizing diverse viral strains. The molecular basis of this neutralization is currently being deciphered by the isolation of HIV-1-neutralizing antibodies. In one infected donor, three neutralizing antibodies, PGT135-137, were identified by assessment of neutralization from individually sorted B cells and found to recognize an epitope containing an N-linked glycan at residue 332 on HIV-1 gp120. Here we use next-generation sequencing and bioinformatics methods to interrogate the B cell record of this donor to gain a more complete understanding of the humoral immune response. PGT135-137-gene family specific primers were used to amplify heavy-chain and light-chain variable-domain sequences. Pyrosequencing produced 141,298 heavy-chain sequences of IGHV4-39 origin and 87,229 light-chain sequences of IGKV3-15 origin. A number of heavy and light-chain sequences of ∼90% identity to PGT137, several to PGT136, and none of high identity to PGT135 were identified. After expansion of these sequences to include close phylogenetic relatives, a total of 202 heavy-chain sequences and 72 light-chain sequences were identified. These sequences were clustered into populations of 95% identity comprising 15 for heavy chain and 10 for light chain, and a select sequence from each population was synthesized and reconstituted with a PGT137-partner chain. Reconstituted antibodies showed varied neutralization phenotypes for HIV-1 clade A and D isolates. Sequence diversity of the antibody population represented by these tested sequences was notably higher than observed with a 454 pyrosequencing-control analysis on 10 antibodies of defined sequence, suggesting that this diversity results primarily from somatic maturation. Our results thus provide an example of how pathogens like HIV-1 are opposed by a varied humoral immune response, derived from intrinsic mechanisms of antibody development, and embodied by somatic populations of diverse antibodies.
Keywords: HIV-1; N-linked glycan; antibody bioinformatics; high-throughput sequencing; immunity.
Figures
References
-
- Archer J., Braverman M. S., Taillon B. E., Desany B., James I., Harrigan P. R., Lewis M., Robertson D. L. (2009). Detection of low-frequency pretherapy chemokine (CXC motif) receptor 4 (CXCR4)-using HIV-1 with ultra-deep pyrosequencing. AIDS 23, 1209–121810.1097/QAD.0b013e32832b4399 - DOI - PMC - PubMed
-
- Boyd S. D., Gaeta B. A., Jackson K. J., Fire A. Z., Marshall E. L., Merker J. D., Maniar J. M., Zhang L. N., Sahaf B., Jones C. D., Simen B. B., Hanczaruk B., Nguyen K. D., Nadeau K. C., Egholm M., Miklos D. B., Zehnder J. L., Collins A. M. (2010). Individual variation in the germline Ig gene repertoire inferred from variable region gene rearrangements. J. Immunol. 184, 6986–699210.4049/jimmunol.1000445 - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources