

Epi-fluorescence microscopy
- PMID: 23026996
- PMCID: PMC3483677
- DOI: 10.1007/978-1-62703-056-4_2
Epi-fluorescence microscopy
Abstract
Epi-fluorescence microscopy is available in most life sciences research laboratories, and when optimized can be a central laboratory tool. In this chapter, the epi-fluorescence light path is introduced and the various components are discussed in detail. Recommendations are made for incident lamp light sources, excitation and emission filters, dichroic mirrors, objective lenses, and charge-coupled device (CCD) cameras in order to obtain the most sensitive epi-fluorescence microscope. The even illumination of metal-halide lamps combined with new "hard" coated filters and mirrors, a high resolution monochrome CCD camera, and a high NA objective lens are all recommended for high resolution and high sensitivity fluorescence imaging. Recommendations are also made for multicolor imaging with the use of monochrome cameras, motorized filter turrets, individual filter cubes, and corresponding dyes being the best choice for sensitive, high resolution multicolor imaging. Images should be collected using Nyquist sampling and images should be corrected for background intensity contributions and nonuniform illumination across the field of view. Photostable fluorescent probes and proteins that absorb a lot of light (i.e., high extinction co-efficients) and generate a lot of fluorescence signal (i.e., high quantum yields) are optimal. A neuronal immune-fluorescence labeling protocol is also presented. Finally, in order to maximize the utility of sensitive wide-field microscopes and generate the highest resolution images with high signal-to-noise, advice for combining wide-field epi-fluorescence imaging with restorative image deconvolution is presented.
Figures

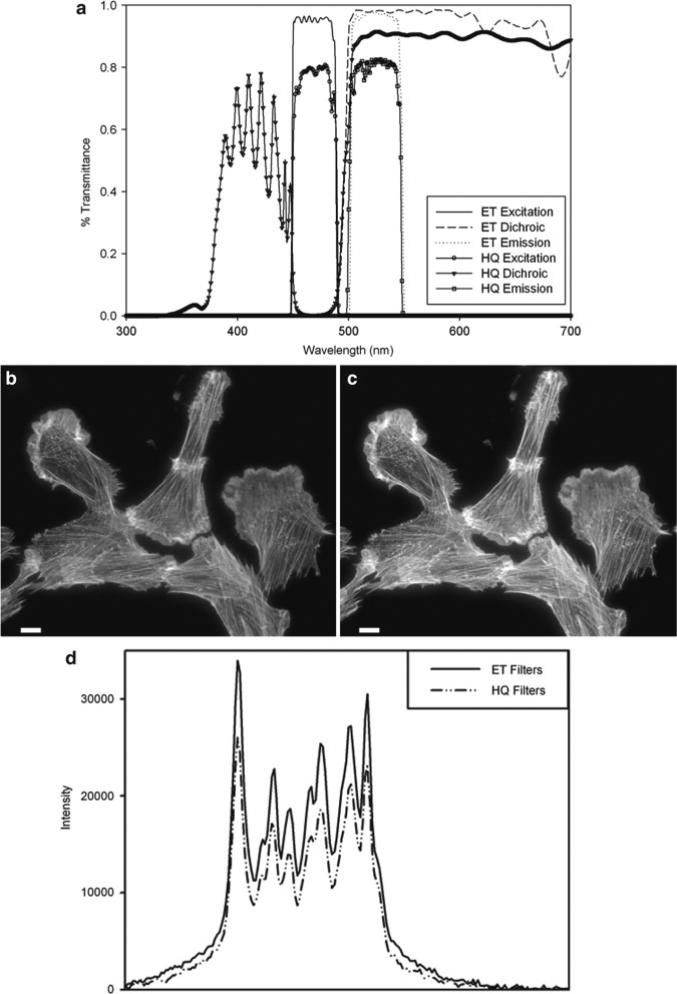

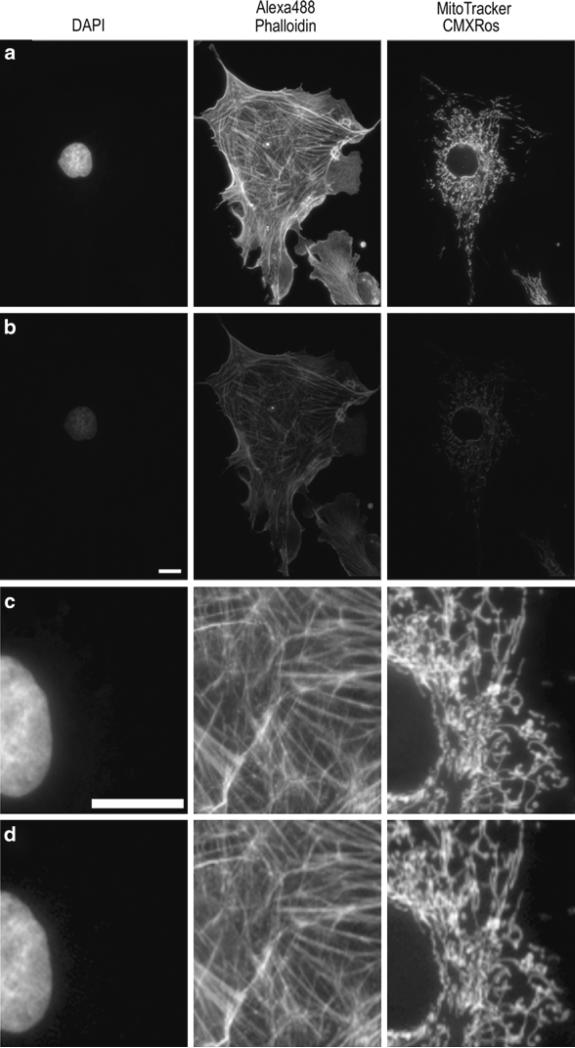



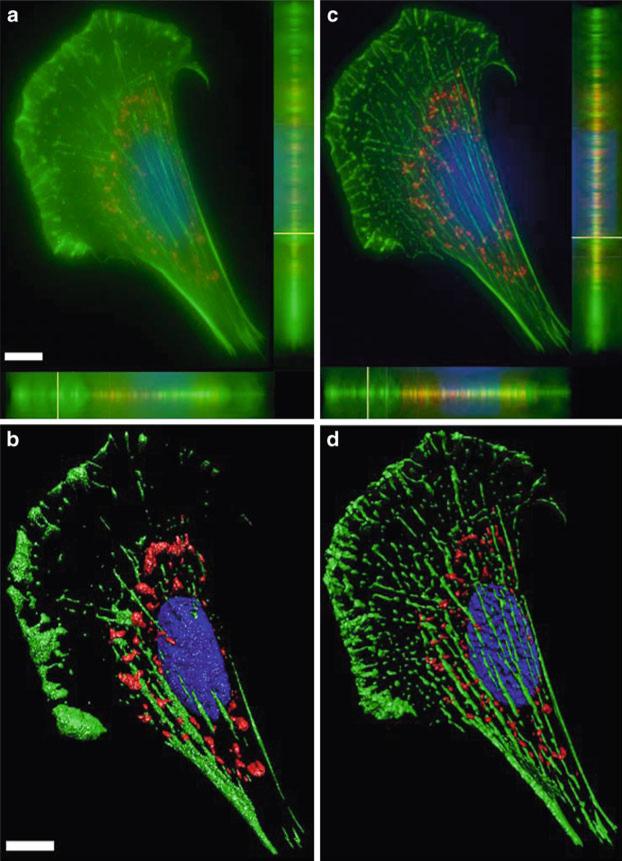
References
-
- Blaydes JP, Vojtesek B, Bloomberg GB, Hupp TR. The development and use of phosphospecific antibodies to study protein phosphorylation. Methods Mol Biol. 2000;99:177–189. - PubMed
-
- DiGiovanna MP, Roussel RR, Stern DF. Production of antibodies that recognize specific tyrosine-phosphorylated peptides. Curr Protoc Cell Biol. 2002 Chapter 16:Unit 16 16. - PubMed
-
- Xu N, Xu M, Zhang YY. Optical detection of single molecules in living cells. Sheng Li Xue Bao. 2005;57:271–277. - PubMed
-
- Lang E, Baier J, Kohler J. Epifluorescence, confocal and total internal reflection microscopy for single-molecule experiments: a quantitative comparison. J Microsc. 2006;222:118–123. - PubMed
-
- Triller A, Choquet D. New concepts in synaptic biology derived from single-molecule imaging. Neuron. 2008;59:359–374. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
