

Neuroprotective efficacy of aminopropyl carbazoles in a mouse model of amyotrophic lateral sclerosis
- PMID: 23027932
- PMCID: PMC3479516
- DOI: 10.1073/pnas.1213960109
Neuroprotective efficacy of aminopropyl carbazoles in a mouse model of amyotrophic lateral sclerosis
Abstract
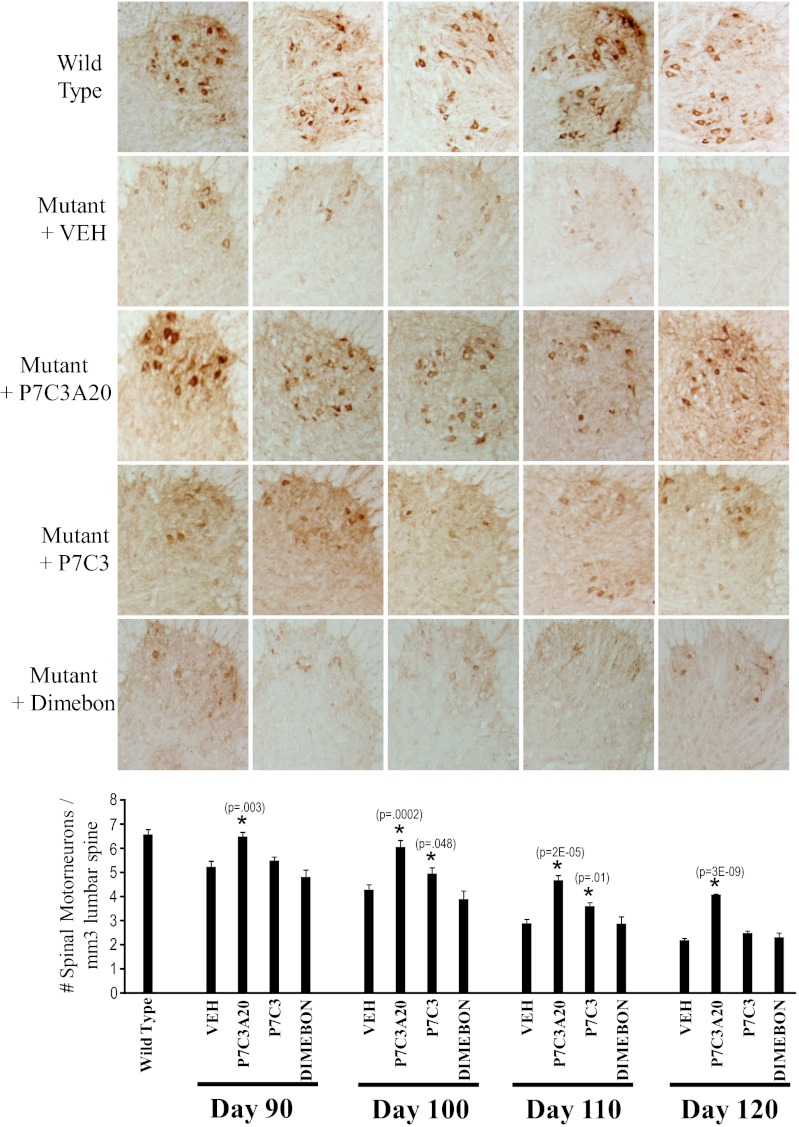
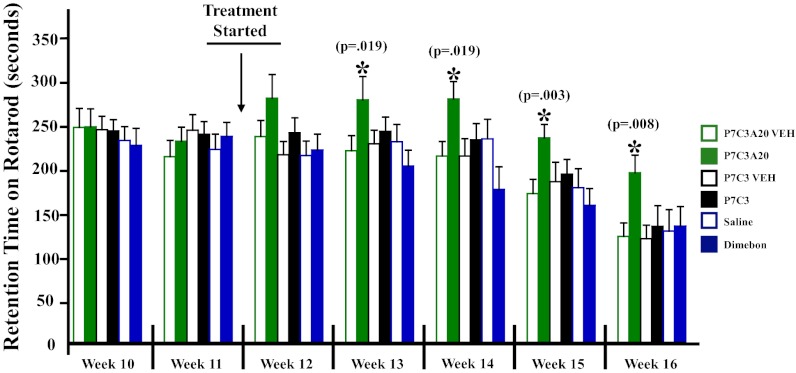
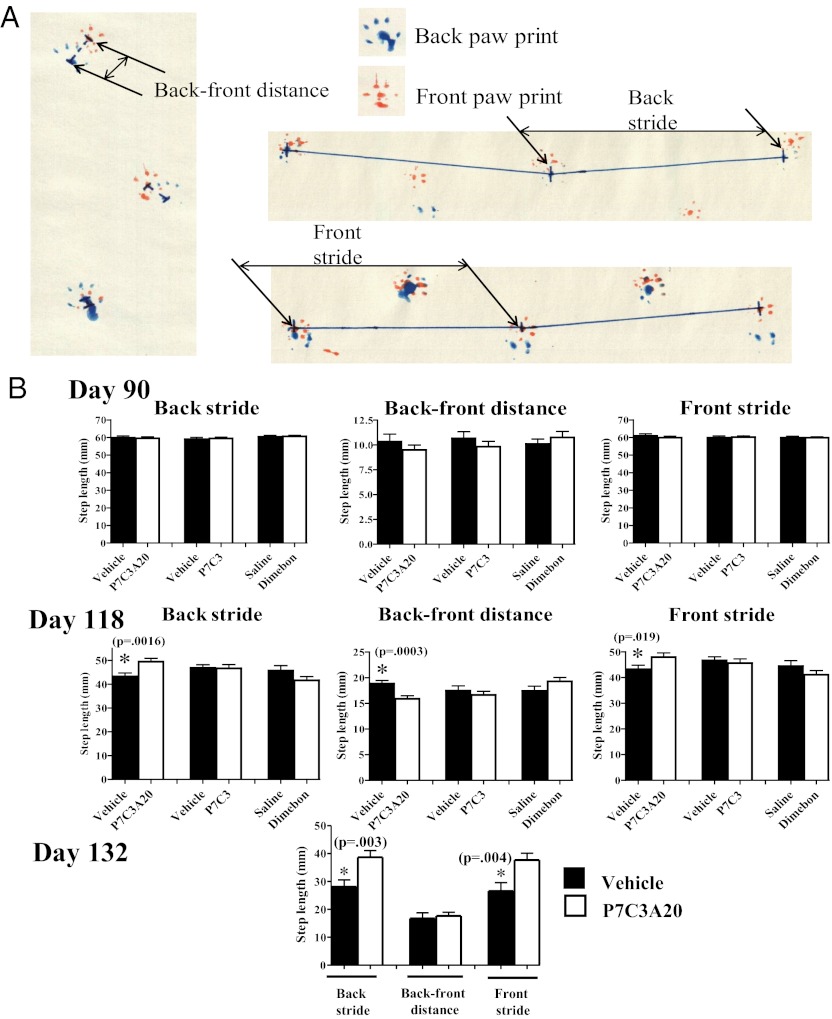
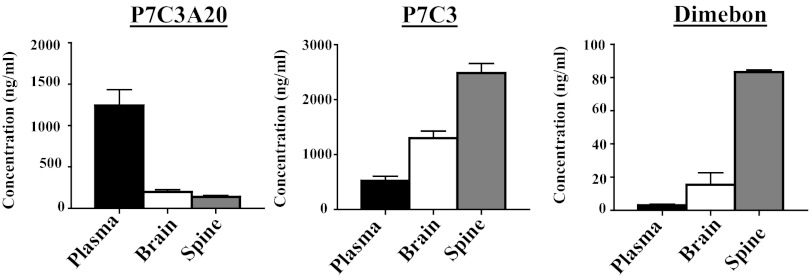
We previously reported the discovery of P7C3, an aminopropyl carbazole having proneurogenic and neuroprotective properties in newborn neural precursor cells of the hippocampal dentate gyrus. We have further found that chemicals having efficacy in this in vivo screening assay also protect dopaminergic neurons of the substantia nigra following exposure to the neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine, a mouse model of Parkinson disease. Here, we provide evidence that an active analog of P7C3, known as P7C3A20, protects ventral horn spinal cord motor neurons from cell death in the G93A-SOD1 mutant mouse model of amyotrophic lateral sclerosis (ALS). P7C3A20 is efficacious in this model when administered at disease onset, and protection from cell death correlates with preservation of motor function in assays of walking gait and in the accelerating rotarod test. The prototypical member of this series, P7C3, delays disease progression in G93A-SOD1 mice when administration is initiated substantially earlier than the expected time of symptom onset. Dimebon, an antihistaminergic drug with significantly weaker proneurogenic and neuroprotective efficacy than P7C3, confers no protection in this ALS model. We propose that the chemical scaffold represented by P7C3 and P7C3A20 may provide a basis for the discovery and optimization of pharmacologic agents for the treatment of ALS.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Comment in
-
Neurodegenerative diseases: Novel route to neuroprotection.Nat Rev Drug Discov. 2012 Dec;11(12):906-7. doi: 10.1038/nrd3899. Nat Rev Drug Discov. 2012. PMID: 23197030 No abstract available.
References
-
- Tandan R, Bradley WG. Amyotrophic lateral sclerosis: Part 1. Clinical features, pathology, and ethical issues in management. Ann Neurol. 1985;18:271–280. - PubMed
-
- Valdmanis PN, Daoud H, Dion PA, Rouleau GA. Recent advances in the genetics of amyotrophic lateral sclerosis. Curr Neurol Neurosci Rep. 2009;9:198–205. - PubMed
-
- Rosen DR, et al. Mutations in Cu/ZN superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;364:59–62. - PubMed
-
- Shaw CE, et al. Familial amyotrophic lateral sclerosis. Molecular pathology of a patient with a SOD1 mutation. Neurology. 1997;49:1612–1616. - PubMed
-
- Valentine JS, Doucette PA, Zittin Potter S. Copper-zinc superoxide dismutase and amyotrophic lateral sclerosis. Annu Rev Biochem. 2005;74:563–593. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
