

High-resolution transcriptome of human macrophages
- PMID: 23029029
- PMCID: PMC3448669
- DOI: 10.1371/journal.pone.0045466
High-resolution transcriptome of human macrophages
Abstract
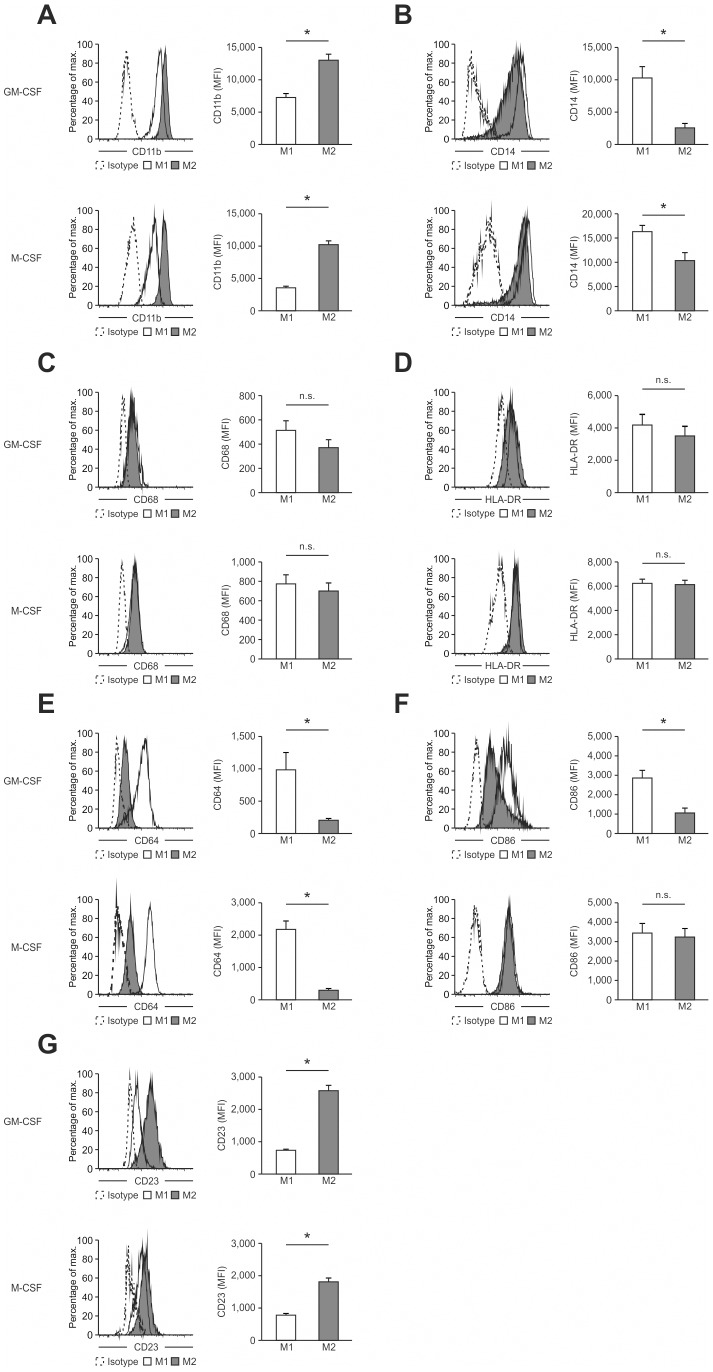
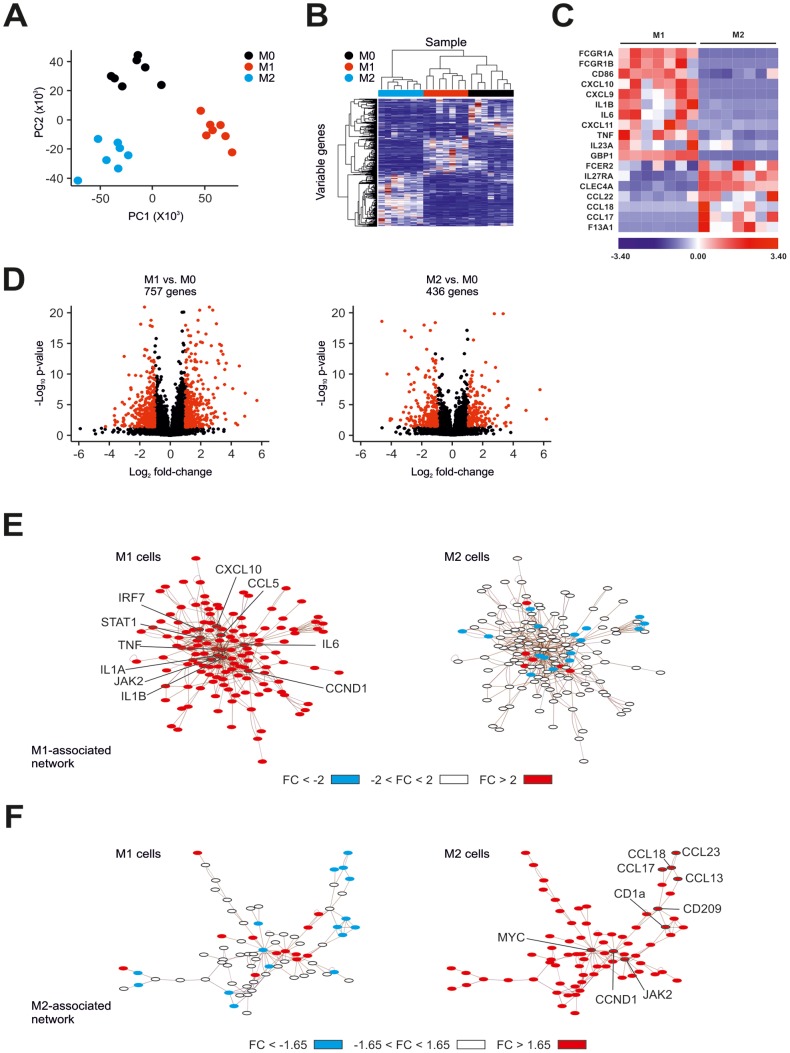
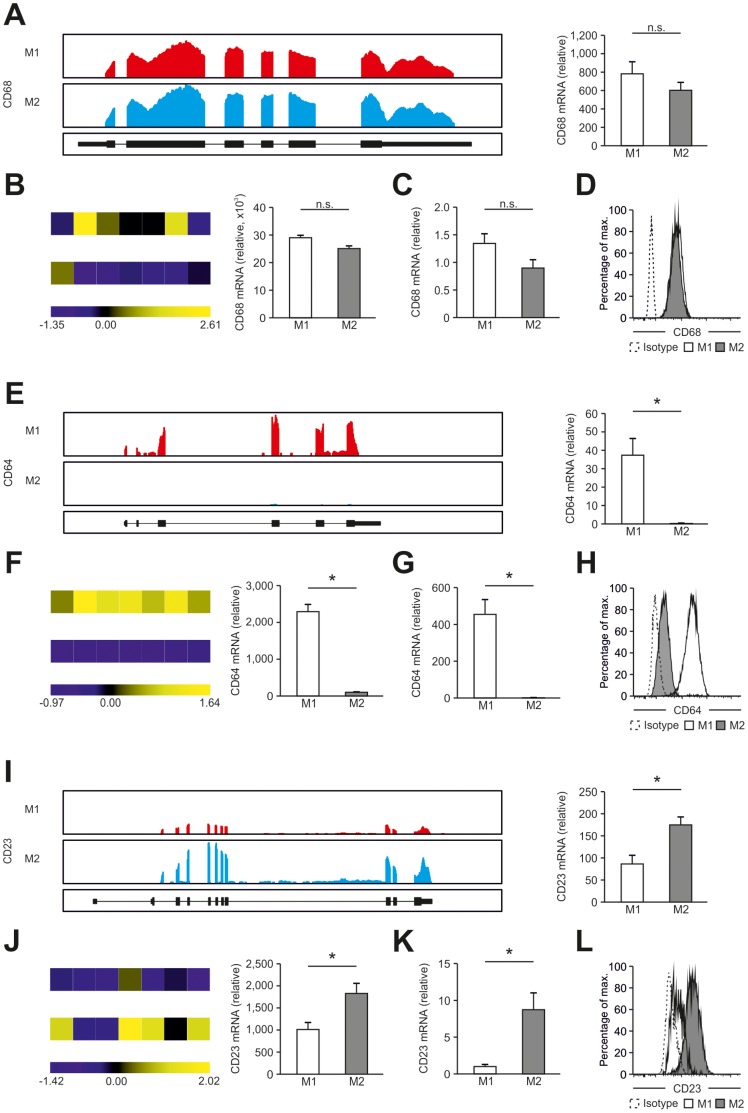
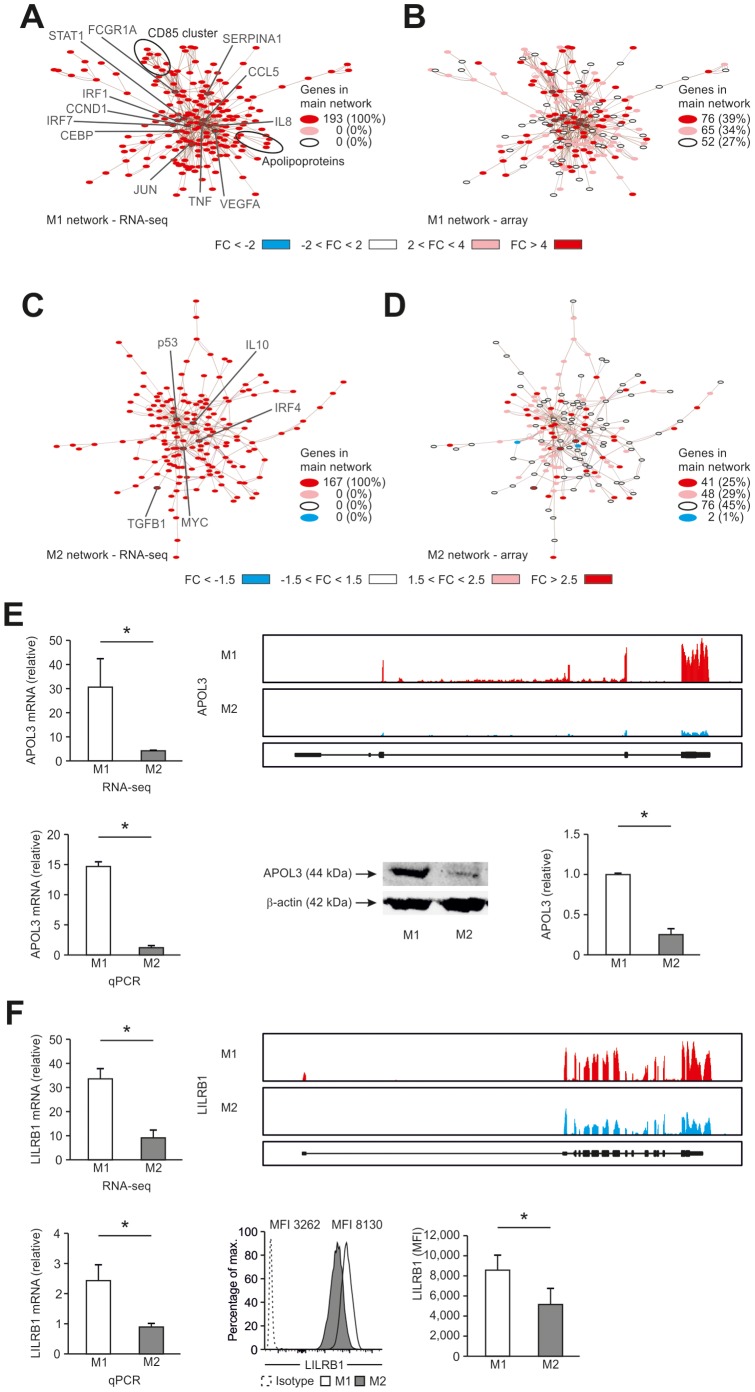
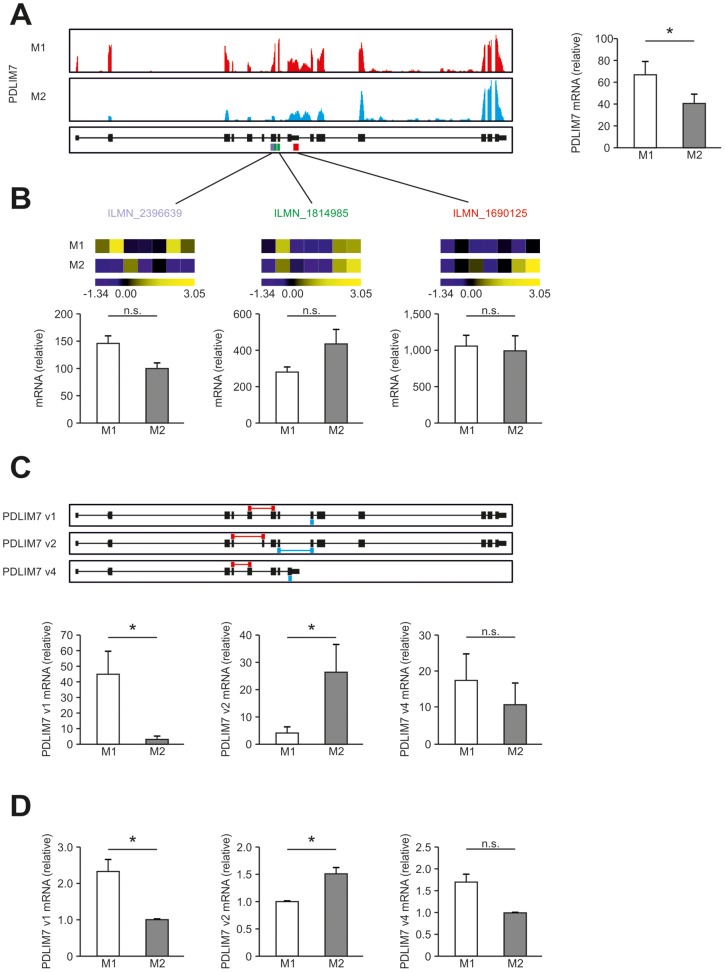
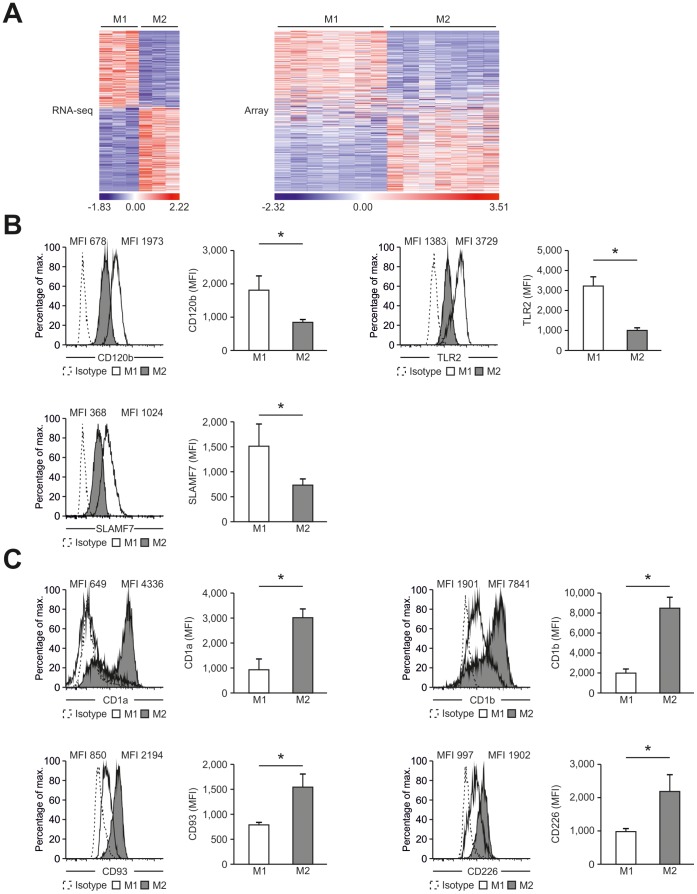
Macrophages are dynamic cells integrating signals from their microenvironment to develop specific functional responses. Although, microarray-based transcriptional profiling has established transcriptional reprogramming as an important mechanism for signal integration and cell function of macrophages, current knowledge on transcriptional regulation of human macrophages is far from complete. To discover novel marker genes, an area of great need particularly in human macrophage biology but also to generate a much more thorough transcriptome of human M1- and M1-like macrophages, we performed RNA sequencing (RNA-seq) of human macrophages. Using this approach we can now provide a high-resolution transcriptome profile of human macrophages under classical (M1-like) and alternative (M2-like) polarization conditions and demonstrate a dynamic range exceeding observations obtained by previous technologies, resulting in a more comprehensive understanding of the transcriptome of human macrophages. Using this approach, we identify important gene clusters so far not appreciated by standard microarray techniques. In addition, we were able to detect differential promoter usage, alternative transcription start sites, and different coding sequences for 57 gene loci in human macrophages. Moreover, this approach led to the identification of novel M1-associated (CD120b, TLR2, SLAMF7) as well as M2-associated (CD1a, CD1b, CD93, CD226) cell surface markers. Taken together, these data support that high-resolution transcriptome profiling of human macrophages by RNA-seq leads to a better understanding of macrophage function and will form the basis for a better characterization of macrophages in human health and disease.
Conflict of interest statement
Figures

References
-
- Martinez FO, Gordon S, Locati M, Mantovani A (2006) Transcriptional Profiling of the Human Monocyte-to-Macrophage Differentiation and Polarization: New Molecules and Patterns of Gene Expression. The Journal of Immunology 177: 7303–7311. - PubMed
-
- Biswas SK, Mantovani A (2010) Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol 11: 889–896. - PubMed
-
- Mantovani A, Sica A (2010) Macrophages, innate immunity and cancer: balance, tolerance, and diversity. Curr Opin Immunol 22: 231–237. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
