

Structural basis for isoform-selective inhibition in nitric oxide synthase
- PMID: 23030042
- PMCID: PMC3835173
- DOI: 10.1021/ar300175n
Structural basis for isoform-selective inhibition in nitric oxide synthase
Abstract
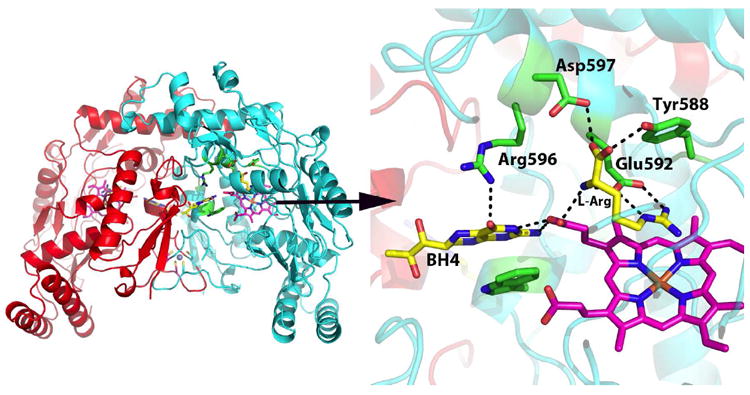
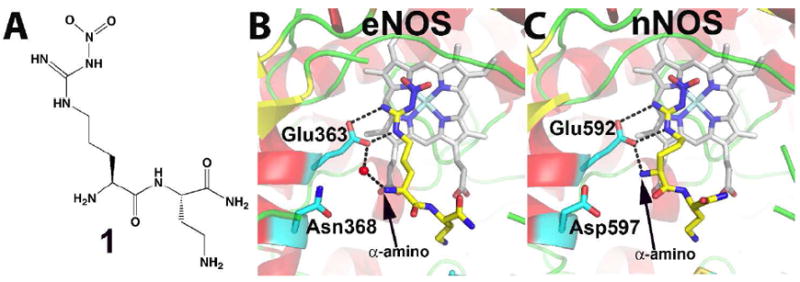
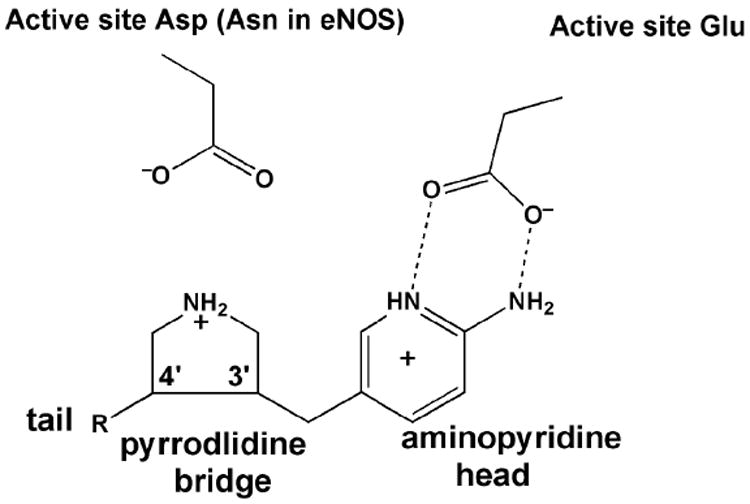
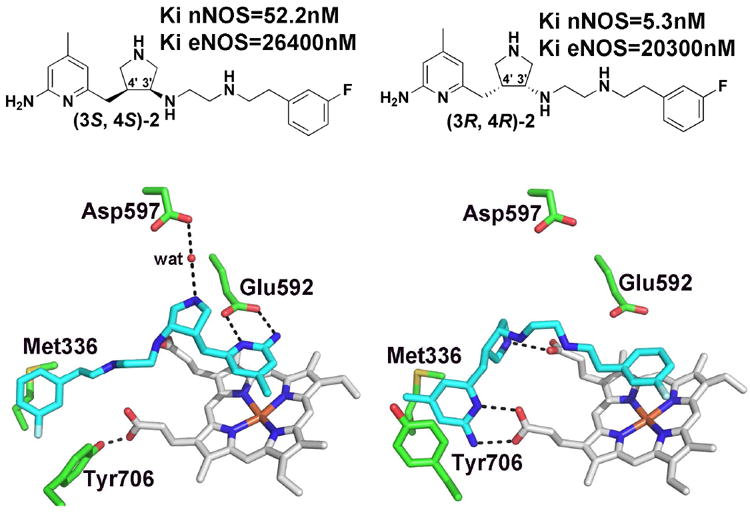
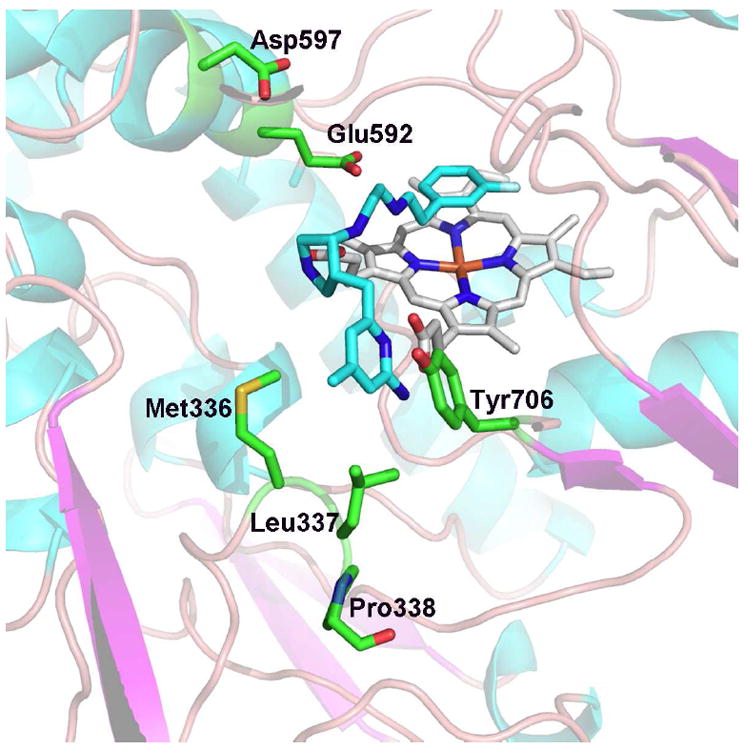
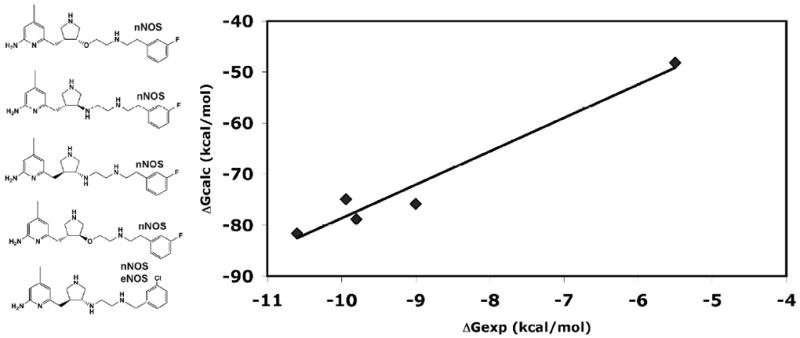
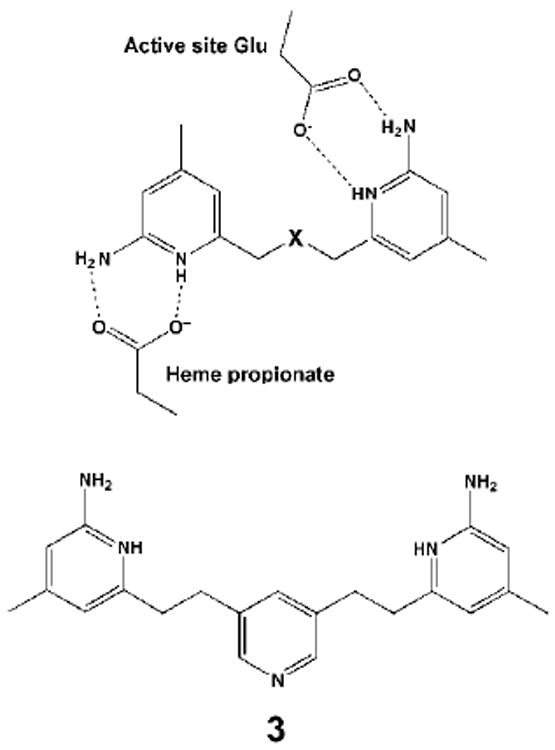
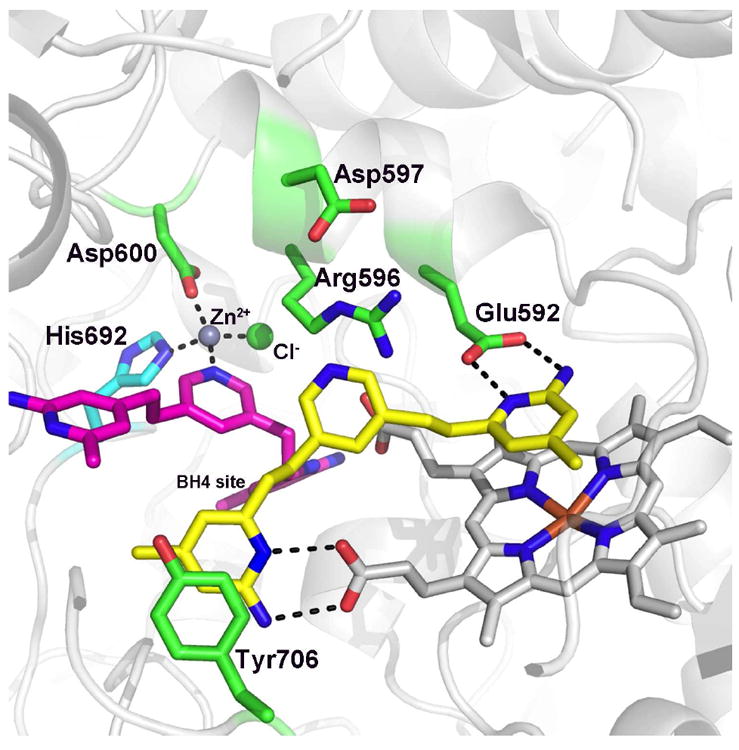
Nitric oxide synthase (NOS) converts l-arginine into l-citrulline and releases the important signaling molecule nitric oxide (NO). In the cardiovascular system, NO produced by endothelial NOS (eNOS) relaxes smooth muscle which controls vascular tone and blood pressure. Neuronal NOS (nNOS) produces NO in the brain, where it influences a variety of neural functions such as neural transmitter release. NO can also support the immune system, serving as a cytotoxic agent during infections. Even with all of these important functions, NO is a free radical and, when overproduced, it can cause tissue damage. This mechanism can operate in many neurodegenerative diseases, and as a result the development of drugs targeting nNOS is a desirable therapeutic goal. However, the active sites of all three human isoforms are very similar, and designing inhibitors specific for nNOS is a challenging problem. It is critically important, for example, not to inhibit eNOS owing to its central role in controlling blood pressure. In this Account, we summarize our efforts in collaboration with Rick Silverman at Northwestern University to develop drug candidates that specifically target NOS using crystallography, computational chemistry, and organic synthesis. As a result, we have developed aminopyridine compounds that are 3800-fold more selective for nNOS than eNOS, some of which show excellent neuroprotective effects in animal models. Our group has solved approximately 130 NOS-inhibitor crystal structures which have provided the structural basis for our design efforts. Initial crystal structures of nNOS and eNOS bound to selective dipeptide inhibitors showed that a single amino acid difference (Asp in nNOS and Asn in eNOS) results in much tighter binding to nNOS. The NOS active site is open and rigid, which produces few large structural changes when inhibitors bind. However, we have found that relatively small changes in the active site and inhibitor chirality can account for large differences in isoform-selectivity. For example, we expected that the aminopyridine group on our inhibitors would form a hydrogen bond with a conserved Glu inside the NOS active site. Instead, in one group of inhibitors, the aminopyridine group extends outside of the active site where it interacts with a heme propionate. For this orientation to occur, a conserved Tyr side chain must swing out of the way. This unanticipated observation taught us about the importance of inhibitor chirality and active site dynamics. We also successfully used computational methods to gain insights into the contribution of the state of protonation of the inhibitors to their selectivity. Employing the lessons learned from the aminopyridine inhibitors, the Silverman lab designed and synthesized symmetric double-headed inhibitors with an aminopyridine at each end, taking advantage of their ability to make contacts both inside and outside of the active site. Crystal structures provided yet another unexpected surprise. Two of the double-headed inhibitor molecules bound to each enzyme subunit, and one molecule participated in the generation of a novel Zn(2+) site that required some side chains to adopt alternate conformations. Therefore, in addition to achieving our specific goal, the development of nNOS selective compounds, we have learned how subtle differences in dynamics and structure can control protein-ligand interactions and often in unexpected ways.
Figures
References
-
- Goodford PJ. Drug design by the method of receptor fit. J Med Chem. 1984;27:558–564. - PubMed
-
- Walkinshaw MD. Protein targets for structure-based drug design. Med Res Rev. 1992;12:317–372. - PubMed
-
- Cushman DW, Cheung HS, Sabo EF, Ondetti MA. Design of potent competitive inhibitors of angiotensin-converting enzyme. Carboxyalkanoyl and mercaptoalkanoyl amino acids. Biochemistry. 1977;16:5484–5491. - PubMed
-
- Wlodawer A, Erickson JW. Structure-based inhibitors of HIV-1 protease. Annu Rev Biochem. 1993;62:543–585. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
