

Novel mutations including deletions of the entire OFD1 gene in 30 families with type 1 orofaciodigital syndrome: a study of the extensive clinical variability
- PMID: 23033313
- PMCID: PMC5497464
- DOI: 10.1002/humu.22224
Novel mutations including deletions of the entire OFD1 gene in 30 families with type 1 orofaciodigital syndrome: a study of the extensive clinical variability
Abstract
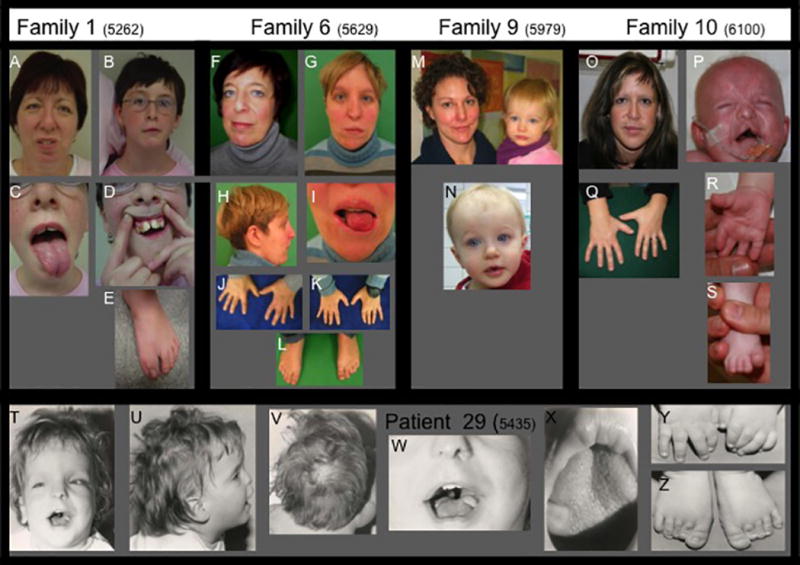
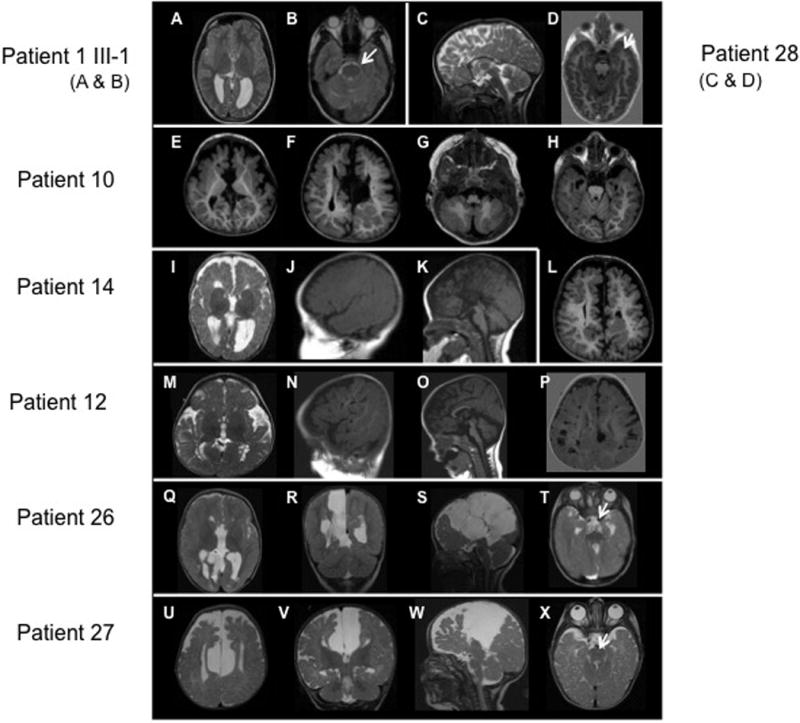
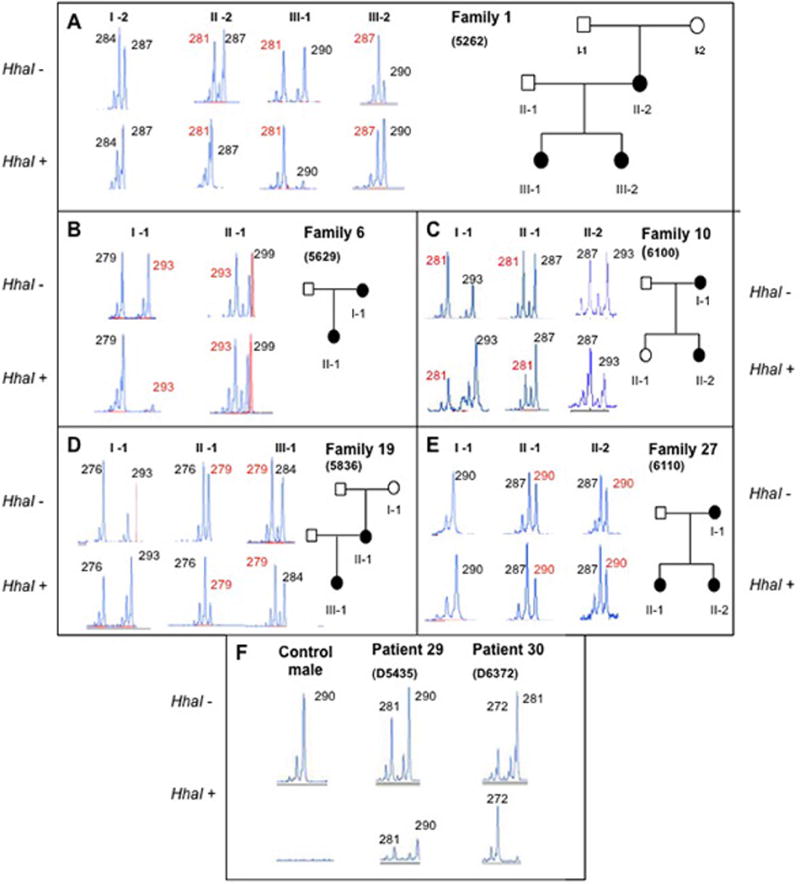
OFD1, now recognized as a ciliopathy, is characterized by malformations of the face, oral cavity and digits, and is transmitted as an X-linked condition with lethality in males. Mutations in OFD1 also cause X-linked Joubert syndrome (JBTS10) and Simpson-Golabi-Behmel syndrome type 2 (SGBS2). We have studied 55 sporadic and six familial cases of suspected OFD1. Comprehensive mutation analysis in OFD1 revealed mutations in 37 female patients from 30 families; 22 mutations have not been previously described including two heterozygous deletions spanning OFD1 and neighbouring genes. Analysis of clinical findings in patients with mutations revealed that oral features are the most reliable diagnostic criteria. A first, detailed evaluation of brain MRIs from seven patients with cognitive defects illustrated extensive variability with the complete brain phenotype consisting of complete agenesis of the corpus callosum, large single or multiple interhemispheric cysts, striking cortical infolding of gyri, ventriculomegaly, mild molar tooth malformation and moderate to severe cerebellar vermis hypoplasia. Although the OFD1 gene apparently escapes X-inactivation, skewed inactivation was observed in seven of 14 patients. The direction of skewing did not correlate with disease severity, reinforcing the hypothesis that additional factors contribute to the extensive intrafamilial variability.
© 2012 Wiley Periodicals, Inc.
Conflict of interest statement
Disclosure statement: The authors declare no financial conflict of interest.
Figures
References
-
- Ahdab-Barmada M, Claassen D. A distinctive triad of malformations of the central nervous system in the Meckel-Gruber syndrome. J Neuropathol Exp Neurol. 1990;49(6):610–20. - PubMed
-
- Bimonte S, De Angelis A, Quagliata L, Giusti F, Tammaro R, Dallai R, Ascenzi MG, Diez-Roux G, Franco B. Ofd1 is required in limb bud patterning and endochondral bone development. Dev Biol. 349(2):179–91. - PubMed
-
- Borozdin W, Boehm D, Leipoldt M, Wilhelm C, Reardon W, Clayton-Smith J, Becker K, Muhlendyck H, Winter R, Giray O, others SALL4 deletions are a common cause of Okihiro and acro-renal-ocular syndromes and confirm haploinsufficiency as the pathogenic mechanism. J Med Genet. 2004;41(9):e113. - PMC - PubMed
-
- Budny B, Chen W, Omran H, Fliegauf M, Tzschach A, Wisniewska M, Jensen LR, Raynaud M, Shoichet SA, Badura M, Lenzner S, Latos-Bielenska A, Ropers HH. A novel X-linked recessive mental retardation syndrome comprisingmacrocephaly and ciliary dysfunction is allelic to oral-facial-digital type I syndrome. Hum Genet. 2006;120:171–178. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
