

Aging effects on DNA methylation modules in human brain and blood tissue
- PMID: 23034122
- PMCID: PMC4053733
- DOI: 10.1186/gb-2012-13-10-r97
Aging effects on DNA methylation modules in human brain and blood tissue
Abstract
Background: Several recent studies reported aging effects on DNA methylation levels of individual CpG dinucleotides. But it is not yet known whether aging-related consensus modules, in the form of clusters of correlated CpG markers, can be found that are present in multiple human tissues. Such a module could facilitate the understanding of aging effects on multiple tissues.
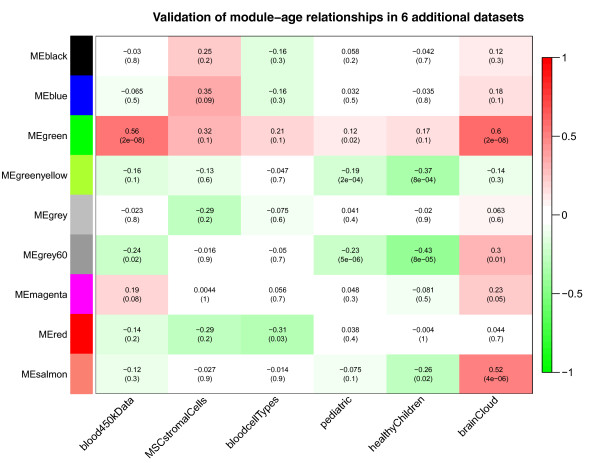
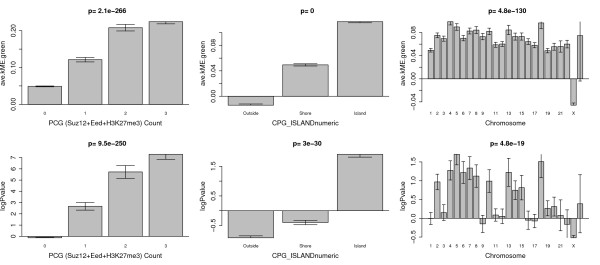
Results: We therefore employed weighted correlation network analysis of 2,442 Illumina DNA methylation arrays from brain and blood tissues, which enabled the identification of an age-related co-methylation module. Module preservation analysis confirmed that this module can also be found in diverse independent data sets. Biological evaluation showed that module membership is associated with Polycomb group target occupancy counts, CpG island status and autosomal chromosome location. Functional enrichment analysis revealed that the aging-related consensus module comprises genes that are involved in nervous system development, neuron differentiation and neurogenesis, and that it contains promoter CpGs of genes known to be down-regulated in early Alzheimer's disease. A comparison with a standard, non-module based meta-analysis revealed that selecting CpGs based on module membership leads to significantly increased gene ontology enrichment, thus demonstrating that studying aging effects via consensus network analysis enhances the biological insights gained.
Conclusions: Overall, our analysis revealed a robustly defined age-related co-methylation module that is present in multiple human tissues, including blood and brain. We conclude that blood is a promising surrogate for brain tissue when studying the effects of age on DNA methylation profiles.
Figures
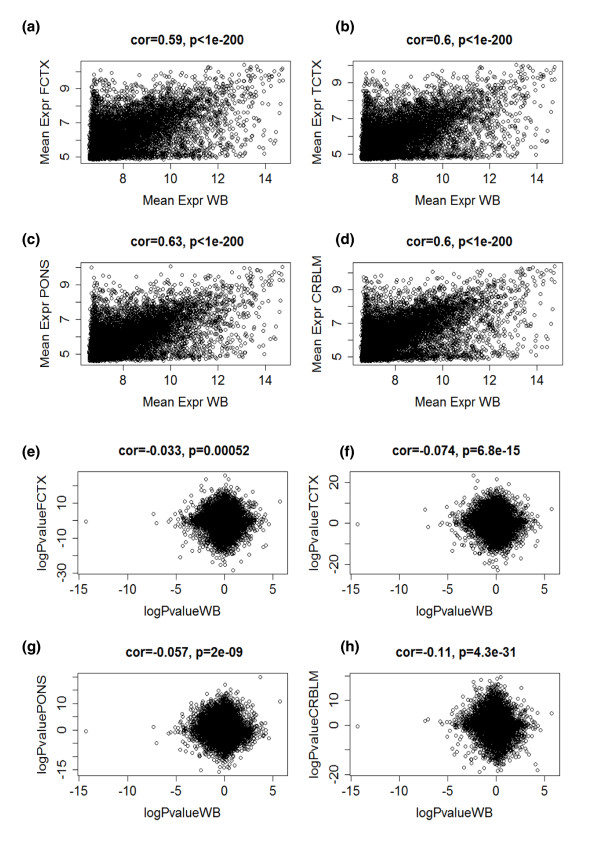
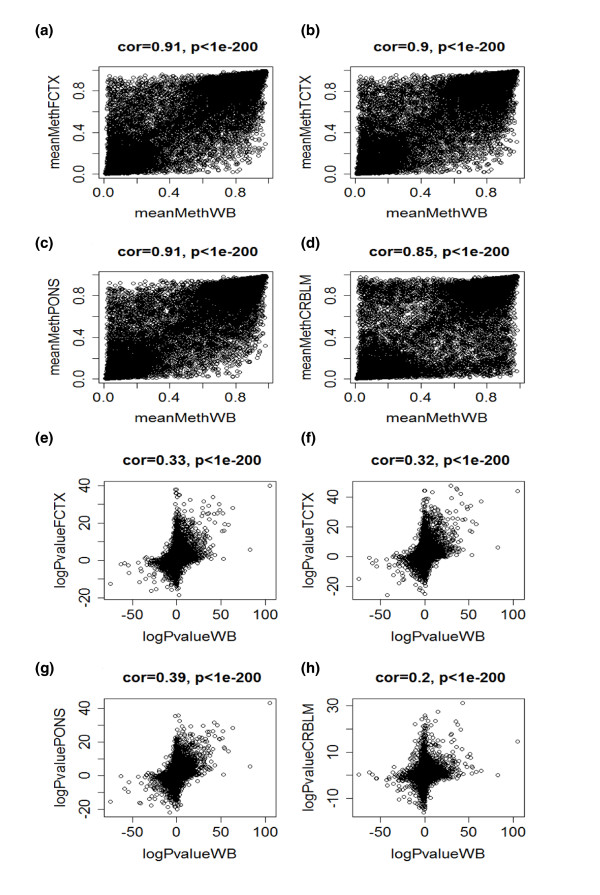
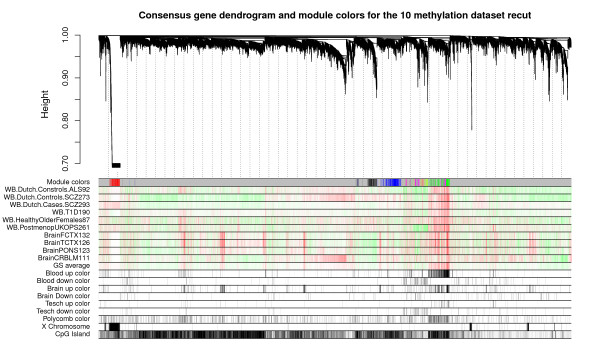
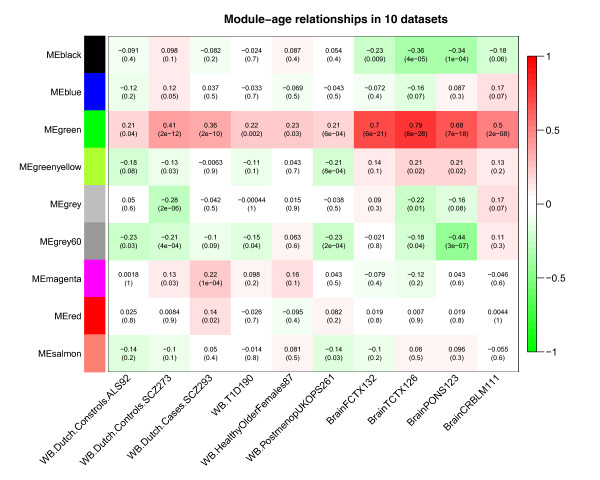
References
-
- Berdyshev G, Korotaev G, Boiarskikh G, Vaniushin B. Nucleotide composition of DNA and RNA from somatic tissues of humpback and its changes during spawning. Biokhimiia. 1967;13:88–993. - PubMed
-
- Bell JT, Tsai P-C, Yang T-P, Pidsley R, Nisbet J, Glass D, Mangino M, Zhai G, Zhang F, Valdes A, Shin S-Y, Dempster EL, Murray RM, Grundberg E, Hedman AK, Nica A, Small KS, Dermitzakis ET, McCarthy MI, Mill J, Spector TD, Deloukas P, The Mu TC. Epigenome-Wide Scans Identify Differentially Methylated Regions for Age and Age-Related Phenotypes in a Healthy Ageing Population. PLoS Genet. 2012;13:e1002629. doi: 10.1371/journal.pgen.1002629. - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
